Chapter 45 Molecular and cellular basis of hypertrophic scarring
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
 IN THIS CHAPTER
IN THIS CHAPTER  PowerPoint Presentation Online
PowerPoint Presentation Online
Introduction
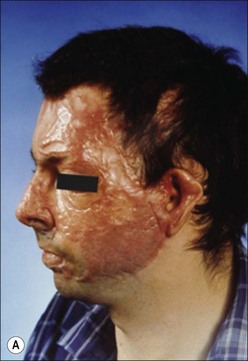
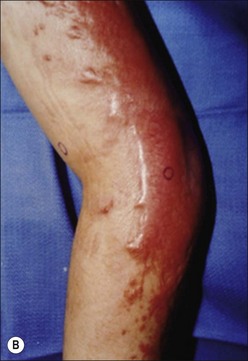
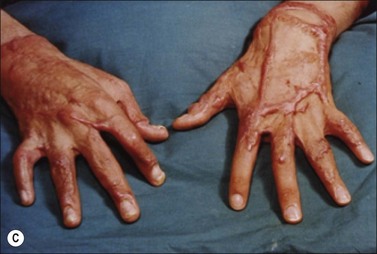
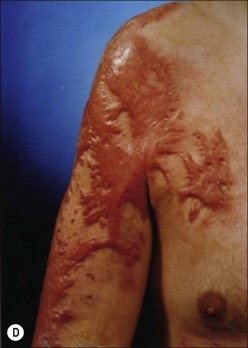
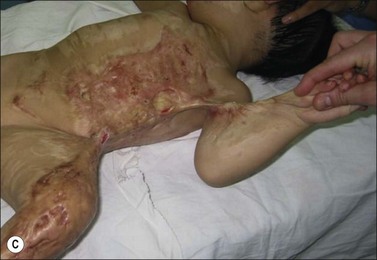
Clinically, post-burn hypertrophic scars (HTS) are elevated, erythematous, pruritic, and inelastic.1 In addition to poor cosmesis, these scars typically form contractures resulting in dysfunction and discomfort, leading to significant morbidity for burn patients (Fig. 45.1). Hypertrophic scar is fundamentally different from normal skin and mature scar in several key ways: 1) the extracellular matrix (ECM) of HTS is significantly altered in both composition and architecture, 2) the behavior of keratinocytes and fibroblasts present in hypertrophic scar is profibrotic compared to mature scar, and 3) many profibrotic cytokines are upregulated and their expression prolonged. Generally, HTS undergoes some remodeling and maturation over time and this may account for the variability in descriptions of HTS. Many features of HTS are shared by other fibroproliferative disorders, including renal fibrosis, pulmonary fibrosis, and scleroderma.2 Thus, understanding the pathophysiology of HTS applies to other areas of medicine, and developments in treating various fibroproliferative diseases can directly affect scar management. Since an exhaustive discussion of every nuance of abnormal wound healing is impossible, we focus on aspects of HTS formation highlighting well understood pathways or novel developments. It is our hope that a deeper understanding of abnormal wound healing pathophysiology and fibrosis will lead to nonsurgical treatments that improve life not only for burn patients, but for the many patients suffering fibrotic diseases.
Extracellular matrix
Extracellular matrix in healing wounds is laid down by fibroblasts and subsequently remodeled as the scar matures. The ECM in HTS displays significant differences from mature scar and normal skin, most notably in the arrangement and composition of collagen bundles, and in the relative proportions of several proteoglycans.3 Owing to the interaction of fibroblasts and ECM these differences not only result from abnormal fibroblast behavior but also contribute to it.4
Collagen
Collagen is the major constituent of ECM, providing a scaffold for cells and mechanical strength to tissues. In HTS the quantity of collagen per unit surface area is increased;5 however, the relative proportion of collagen in HTS is decreased compared to normal skin owing to much greater increases in proteoglycans and glycoproteins.3 In normal skin the majority of collagen is type I (80%) with smaller amounts of type III (10–15%) and type V (minimal) resulting in thick, regular collagen fibril bundles running parallel to the skin surface.6 In contrast, HTS is composed of greatly increased amounts of type III (33%)7 and type V (10%)8 collagen, which drastically alters collagen fibrils making them thinner6 and disorganized.9 In normal wound healing, type III collagen appears early then gradually disappears as the scar is remodeled and matures,10 but this does not occur in HTS where persistently high levels reflect biological immaturity.11
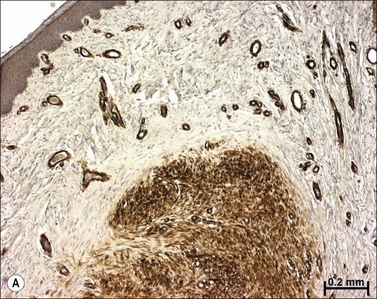
Classic histologic descriptions of HTS highlight ‘whorls’ and ‘nodules’ of poorly organized collagen encapsulated in more normal-appearing collagen fibrils,6 as seen in Figure 45.6. However, not only are collagen fibril composition and morphology altered in HTS, but interfibrillar spacing is also irregular and greatly increased.12 This space is filled with proteoglycans and glycoproteins, whose composition is markedly different from normal skin and mature scar.
Proteoglycans and glycoproteins
Proteoglycans are formed by a protein core, often with repeating units such as leucine in decorin, and glycosaminoglycan side chains.13 These side chains are ionized and hydrophilic, and thus mainly responsible for tissue water retention.14 Early studies of HTS demonstrated elevated glycosaminoglycan levels,15 which are responsible for the hyperhydrated state of HTS leading to its classically increased turgor. These levels of glycosaminoglycan are not uniformly elevated. Instead, certain proteoglycans are downregulated and others upregulated, with unique implications for HTS.
Decorin is a prototypical small, leucine-rich proteoglycan (SLRP) produced in abundance in normal skin and mature scar but reduced by 75% in HTS.3 Originally named for ‘decorating’ collagen fibrils, decorin affects wound healing via several distinct and complementary pathways. Decorin binds to collagen fibrils, controlling their diameter, morphology,16 and interfibrillar distance.17 In decorin knockout mice collagen fibrils are irregular in morphology and have highly variable diameters.18 Decorin binds to and inactivates the profibrotic cytokines transforming growth factor-β (TGF-β),19 and platelet-derived growth factor (PDGF).20 The effects of this inactivation are most readily visible in fibroblast-populated collagen lattices where decorin significantly reduces contraction by normal and HTS fibroblasts.21,22 Decorin also binds to and antagonistically downregulates several cell surface receptor tyrosine kinases: EGFR,23 HGFR,24 and IGF1R,25 which reduces cellular proliferation and migration. Decorin production increases significantly as scars mature.26 In a mouse model of diabetes, renal fibrosis and nephropathy developed significantly faster in decorin knockout than in wild-type mice.27 Similarly, upregulating decorin production via an adenoviral vector in mouse models of bleomycin-induced pulmonary fibrosis reduced fibrosis.28
In contrast to decorin downregulation, two other proteoglycans, biglycan, and versican, are significantly upregulated in HTS. Biglycan is 57% similar to decorin in amino acid sequence but with two dermatan sulfate chains, and is believed to have originated as a gene duplication of decorin.29 Despite these similarities, biglycan and decorin have significantly different functions in vivo. Biglycan is minimally present in normal skin but significantly upregulated in fibrosis, yet does not compensate for the lack of decorin.29,30 Versican is also significantly upregulated in HTS, up to six times higher than in normal skin,3 where it is normally confined to the proliferating epidermis.31 As a large proteoglycan with 12–30 glycosaminoglycans chains, versican is most likely responsible for the increased hydration and turgor leading to the increased volume of HTS.5
The most common glycoprotein in ECM is fibronectin, which has effects on cell–matrix interaction via its interaction with integrins. Although the role of fibronectin in HTS is unclear, its upregulation in HTS,32 influence on the assembly of other ECM proteins, and interaction with cellular integrins33 suggest it too plays a role in fibroblast behavior and HTS formation.
Cellular contributions to hypertrophic scar
Hypertrophic scar fibroblasts
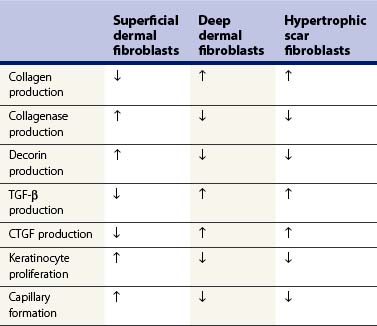
Fibroblasts are the cells primarily responsible for ECM production and remodeling in wound healing. Numerous studies have demonstrated that dermal fibroblasts can be divided into distinct subpopulations: superficial (papillary), and deep (reticular), based on both physical location and phenotype.34–36 When characteristics of superficial dermal, deep dermal, and HTS fibroblasts are compared, as in Table 45.1, it becomes clear that HTS fibroblasts most closely resemble deep dermal fibroblasts.36 These in vitro results correlate directly with a clinically relevant dermal scratch model developed by Dunkin and colleagues.37 In this model a linear skin wound is created with depth that increases along its length from no injury to full-thickness penetration. This results in regeneration in the superficial portion (depth ≤0.56 mm) with minimal scar; whereas HTS formation occurs in the deeper portion of the wound.37 Combining these basic science and clinical observations with the tendency of HTS to form following deep partial- to full-thickness burns suggests two possible theories of HTS formation: 1) selective proliferation of deep dermal fibroblasts after fibrogenic cytokine stimulation, or 2) destruction of superficial dermal fibroblasts by thermal injury, leaving only deep dermal fibroblasts to repopulate the wound.38 Both theories are consistent with an experimental model where human skin was grafted onto the backs of nude mice and subsequently injured. In this model, deep dermal fibroblasts closed wounds and superficial dermal fibroblasts then remodeled them.39 It is possible that in deep burns superficial fibroblasts are not present to initiate this remodeling process, leaving the healing wound in a hyperactive state (Fig. 45.2).

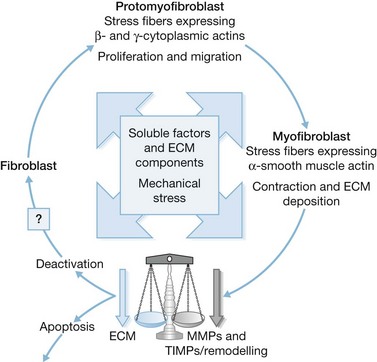
Role of myofibroblasts in normal and pathological situations
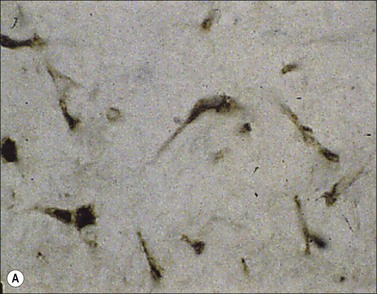
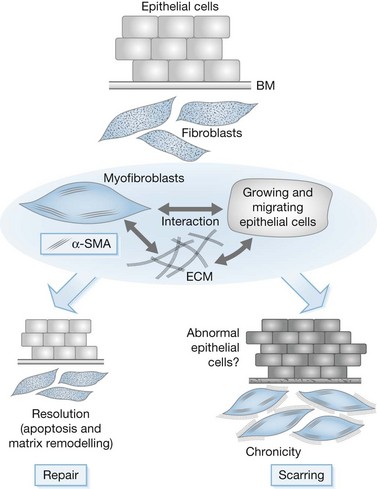
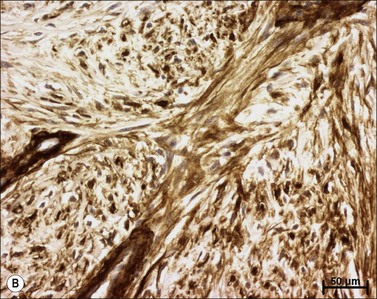
Myofibroblasts are cells that have acquired a phenotype intermediate between fibroblasts and smooth muscle cells. Presently, it is accepted that myofibroblast modulation of fibroblastic cells begins with the appearance of precursor proto-myofibroblasts, whose stress fibers contain only β- and γ-cytoplasmic actins. These proto-myofibroblasts acquire de novo contractile bundles whose stress fibers generate sufficient forces to pull cells forward to populate tissue spaces in a migration process and to pre-remodel the ECM. Proto-myofibroblasts evolve, but not necessarily always, into the differentiated myofibroblast.40 Fully differentiated myofibroblasts express α-smooth muscle actin (α-SMA), the actin isoform present in typical contractile vascular smooth muscle cells (Fig. 45.3). The presence of α-SMA is directly related to the contractile activity of myofibroblasts. A direct correlation has been demonstrated both in vitro and in vivo between the level of α-SMA expression and myofibroblast contraction.41,42 Myofibroblasts also exhibit some similarities with pericytes.43 In physiological conditions, after healing, myofibroblasts undergo apoptosis44 and only a few fibroblasts are left to ensure renewal of the ECM.
Among the soluble factors, TGF-β1 is a potent inducer of myofibroblast differentiation.45,46 TGF-β1 action on myofibroblast differentiation is only possible in the presence of ED-A fibronectin, which underlines the fact that ECM components play an important role in soluble factor activity.48 Granulocyte–macrophage colony-stimulating factor stimulates macrophage proliferation and myofibroblast differentiation, thereby promoting granulation tissue formation.49,50 Endothelin has also a positive effect on differentiation and activation of myofibroblasts. This peptide also induces myofibroblast contraction and migration.51 More recently, it has been shown that granulation tissue formation is modified by chemical denervation.52 This peripheral nervous system involvement in tissue repair has likewise been shown in the liver; in this organ, in an experimental model of fibrosis using carbon tetrachloride treatment, chemical denervation significantly reduces matrix deposition and myofibroblast differentiation.53
Role of mechanical stress and myofibroblasts
Myofibroblast cells, because of their contractile properties and privileged relationships with ECM, can modify their activity depending on the mechanical environment. It has been shown, in gingival fibroblasts, that α-SMA expression induced by TGF-β1 is regulated by the compliance of collagen gels on or in which they are cultured.54 The direct effects of mechanical stress on fibroblasts can be easily shown in culture using stressed fibroblast-populated collagen lattices (Fig. 45.4). Moreover, myofibroblast differentiation features, such as stress fibers, ED-A fibronectin or α-SMA expression, appear earlier in granulation tissue subjected to increased mechanical tension by splinting of the full-thickness wound with a plastic frame than in normally healing wounds.42 It has also been shown that fibroblasts cultured on substrates of variable stiffness present different phenotypes. Cultured fibroblasts do not express stress fibers on soft surfaces, but when the stiffness of the substrate increases a sudden change in cell morphology occurs and stress fibers appear.55,56 Shear forces exerted by fluid flow are also able to induce TGF-β1 production and differentiation of fibroblasts cultured in collagen gels in the absence of other external stimuli, such as cytokine treatment.57 Intercellular mechanical coupling of stress fibers via adherens junctions improves contraction of collagen gels by myofibroblasts.58 By assessing spontaneous intracellular Ca2+ oscillations, Follonier et al. have shown that intracellular Ca2+ oscillations are coordinated between contacting myofibroblasts via adherens junctions, but randomly between fibroblasts and non-contacting cells.59 They propose the following model of mechanical coupling for myofibroblasts: individual cell contraction is transmitted via adherens junctions and leads to opening of mechanosensitive ion channels in adjacent cells. The resulting Ca2+ influx induces a contraction that can feed back on the first cell and/or stimulate other contacting cells working like a syncytium. This mechanism could improve the remodeling of cell-dense tissue by coordinating the activity of myofibroblasts.60


Pathological repair (hypertrophic scars and keloids)
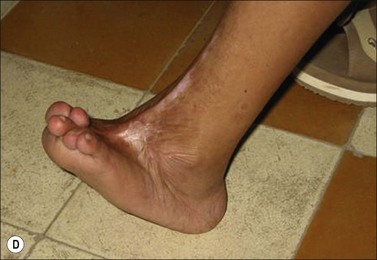
Pathological wound healing can result from impaired remodeling of the granulation tissue, leading to abnormal cutaneous repair in hypertrophic or keloid scars (Fig. 45.5). Keloid and HTS differ in their expression of α-SMA; indeed, in keloids, no α-SMA is observed, although proto-myofibroblasts could account for large amounts of extracellular matrix but are unable to contract, whereas numerous myofibroblasts express this protein in HTS, explaining the fact that contracture often appears specifically in HTS.38,61 Thus, the use of α-SMA to differentiate HTS and keloids has been proposed.62 The presence of contractile myofibroblasts in HTS is responsible for the formation of contractures that interfere with function and may require extensive reconstructive surgery (Fig. 45.6). Moreover, keloids contain thick collagen fibres, whereas HTS contain thin fibres organized in nodules.61,63 It emphasizes that the different processes involved in collagen maturation together with the effects of the matrix metalloproteinase (MMP)/tissue inhibitor of MMP(TIMP) system play an important role in excessive scar formation. The expression of lysyl hydroxylase (LH)-2b, a splice variant of LH-2, an enzyme involved in the cross-linking of the collagen fibrils, has been linked to fibrotic development occurring in pathologic situations.64 Animal models mimicking this specific expression of LH-2b are available and could be used to test new anti-scarring therapies based on the inhibition of LH-2b.65 In these lesions, the normal healing process cannot be achieved and granulation tissue continues to grow, owing to an abnormal and excessive secretion of growth factors and/or lack of molecules inducing apoptosis or ECM remodeling. Interestingly, HTS have an excess of microvessels, most of which are partially or totally occluded due to a functional regression of endothelial cells induced by (myo)fibroblast hyperactivity.66 In excessive scarring a focal upregulation of p53 expression, which probably inhibits apoptosis, has been observed. Extracellular matrix modifications also seem to be an important factor in induction of the apoptotic process. In vivo, covering granulation tissue by a vascularized skin flap induces an upregulation of MMP together with a decrease in TIMP, leading to a rapid loss of granulation tissue cells by apoptosis.67 In vitro, the matrix environment can modulate fibroblast apoptosis. Furthermore, in HTS, the mechanical forces obtained by compression of the scar are able to restore the classic organization observed in normal wounds and trigger the disappearance of myofibroblasts by apoptosis.68 Thus, mechanical stress can maintain myofibroblast differentiation. It has been shown that mechanical loading early in the proliferative phase of wound healing produces HTS by inhibiting cellular apoptosis.69 In contrast, it is suggested that mechanical challenge could be a clinically relevant strategy to improve ischemic and chronic wound healing by supporting myofibroblast formation.70 Interestingly, oncogenic-Ras-transformed human fibroblasts lose growth factor selectivity and cell matrix density-dependent inhibition of migration.71 In the liver, and certain other organs, stiffness appears to result from matrix cross-linking and possibly other unknown variables in addition to matrix quantity, suggesting that increased stiffness may play an important role in initiating the early stages of fibrosis.72 The epithelium could also be involved in the development of excessive scarring.73 Hakvoort et al. have shown that, in HTS, keratinocytes expressed an activated CD36+ phenotype.74 They suggest that HTS formation is not only due to dermal dysfunction, but is the result of a perturbation affecting dermal–epidermal interactions involving neurohormonal factors. Indeed, a ‘neurogenic inflammation hypothesis’ has been suggested.75 Mechanical stress stimulates mechanosensitive nociceptors in skin sensory fibers, which release neuropeptides involved in vessel modifications and fibroblast activation.
Origin of (myo)fibroblasts
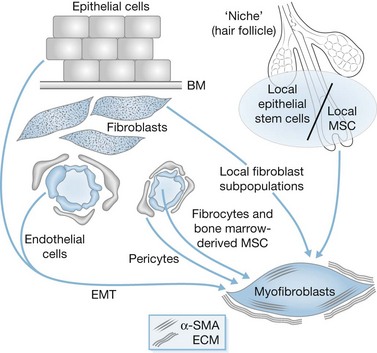
It is now accepted that myofibroblasts can originate from various cell types, as illustrated in Figure 45.7. The majority of these cells originate from local recruitment of connective tissue fibroblasts. For example, in skin, dermal fibroblasts located in the edges of the wound can acquire a myofibroblast phenotype and participate in tissue repair.76,77 In diffuse cutaneous systemic sclerosis, microvascular pericytes constitute a cellular link between microvascular damage and fibrosis by transdifferentiating into myofibroblasts.78 In the liver, the role of hepatic perisinusoidal hepatic stellate cells has been widely studied and their key role during fibrogenesis has been clearly demonstrated.79 Recently, it has also been shown that portal fibroblasts are involved in the formation of portal septa.80 Vascular smooth muscle cells residing in the walls of portal vein branches and portal arteries have been implicated in fibrosis observed in chronic schistosomiasis.81 In the kidney, both mesangial cells and interstitial fibroblasts of the medulla can acquire a myofibroblast phenotype and participate in ECM deposition after damage.82,83 Moreover, the involvement in tissue repair of local mesenchymal stem cells is becoming better understood. These progenitor cells have been described in the dermal sheath that surrounds the outside of the hair follicle facing the epithelial stem cells, constituting a niche of stem cells. They are involved in the regeneration of the dermal papilla and can also become wound healing (myo)fibroblasts after an injury.84 This concept of a cell association, able to reconstitute the different organ cell populations and constituting a niche of stem cells, is currently discussed in diverse organs, notably the liver, in the periportal zone containing Hering’s canals.85,86 Recent data have shown the implication of circulating cells, called fibrocytes, in the tissue repair process.90 Another type of circulating cell originating from bone marrow participates in tissue repair. These mesenchymal stem cells are bone marrow-derived non-hematopoietic precursor cells91,92 which contribute to the maintenance and regeneration of connective tissues through engraftment. Indeed, they have the capacity to engraft into several organs and to differentiate into wound healing myofibroblasts. However, recently it has been suggested that fibroblasts/myofibroblasts that participate in cutaneous wound healing are not derived from circulating progenitor cells.93 Finally, epithelial- and endothelial-to-mesenchymal transition (EMT), a process by which differentiated (or malignant) epithelial cells and endothelial cells undergo a phenotypic conversion that gives rise to the matrix-producing fibroblasts and myofibroblasts, is increasingly recognized as an integral part of tissue fibrogenesis after injury, particularly in the kidney94 and during stroma reaction formation. However, the degree to which this process contributes to fibrosis and stroma reaction in the skin remains a matter of intense debate and is likely to be context dependent. Altogether, mesenchymal stem cells, fibrocytes, bone marrow-derived cells and cells derived from EMT, may represent alternative sources of myofibroblasts when local fibroblasts are not able to respond. These diverse cell types probably contribute to the appearance of myofibroblast subpopulations whose phenotype can be modulated by their interactions with neighboring cells and ECM.95,96