Figure 1: Main discoveries in genetics.
b. The concept of RNA interference
Epigenetics was, until the end of the 1980s, a topic related only to chemical modifications of DNA or proteins, allowing transcriptional repression or activation. However, in the early 1990s, several studies reported RNA-mediated inhibition of protein expression, also called gene-silencing, occurring at the post-transcriptional level; which is related to epigenetics regulation of gene expression. First observed in plants by Richard Jorgensen and colleagues (Napoli et al. 1990), gene silencing was also described in the mold Neurospora crassa (Romano and Macino 1992), and named “quelling.” These seminal observations introduced the concept of post-transcriptional gene silencing (PTGS) or RNA silencing (Figure 2).
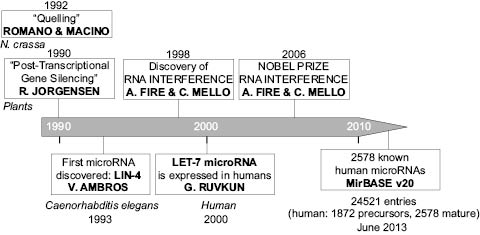
Figure 2: Timeline of the discoveries in microRNA research.
In 1998, Fire and Mello observed in the worm Caenorhabditis elegans that double-stranded RNA (dsRNA) was capable of triggering sequence-specific inhibition of protein expression, a phenomenon that they called “RNA interference” (Fire et al. 1998). This term encompasses the post-transcriptional inhibition of gene expression by two types of small RNA molecules: small interfering RNAs (siRNAs) and microRNAs. Both siRNAs and microRNAs are small double-stranded RNAs. They differ in that siRNAs arise from long exogenous RNA precursors such as viruses, transposons ,or plasmids taken up by cells (they can also be applied on cells as synthetic siRNAs), while microRNAs are transcribed from the cellular genomic DNA and further processed.
MicroRNAs are small double-stranded noncoding RNAs (20–25 nucleotides in length). The first microRNA, Lin-4, was discovered in the worm Caenorhabditis elegans in 1993 by Victor Ambros and colleagues (Lee et al. 1993) and the first human one, Lethal-7 or Let-7, was discovered in 2000 by Gary Ruvkun and collaborators (Reinhart et al. 2012; Pasquinelli et al. 2000) (Ref Figure 2). The subsequent development of miR cloning alowed the identification of microRNA homologs in many vertebrate species, demonstrating the evolutionarily conservation of the major microRNAs across many species.
In only thirteen years, known human microRNA precursors reported in MirBASE, reached the number of 1872 (corresponding to 2578 mature microRNAs) (MirBASE v10, June 2013), giving evidence of the high interest of the scientific community for this new type of cellular components. The future elucidation of the entire human repertoire of microRNAs will be an important step in fully understanding their wide range of functions.
The discovery of RNA-mediated post-transcriptional regulatory mechanisms and the contribution to biology and medicine are so important that in 2006, the Nobel Prize in Physiology or Medicine was awarded to Andrew Z. Fire and Craig C. Mello for the discovery of RNA interference (Ref Figure 2), opening the way to breakthrough technologies to study the effects of the loss of function of specific genes in mammalian cells (Almeida et al. 2011) and to the use of these technologies in clinical therapeutics (Sindhu et al. 2012).
4.3.3.2 MICRORNAS – NOMENCLATURE, STRUCTURE, FUNCTION, MECHANISM OF ACTION
MiRNAs follow a particular nomenclature, allowing their identification in databases such as MirBASE, and insuring homogeneous principles of naming for the scientific community (Kozomara and Griffiths-Jones, 2014). The following are the main nomenclature rules for microRNAs. Hence, a sequential numerical identifier is assigned to each newly discovered microRNA: miR-29, miR-30, miR-31, etc. Usually, the mature microRNA sequences are designated “miR,” whereas the precursors and genes are labeled “mir.” The complete name of a microRNA should use a three- to four-letter code to indicate the species. Homo sapiens species, for example, will be indicated by the letters “hsa” (e.g., hsa-miR-29). In mouse, the prefix will be “mmu” (Mus musculus), whereas for drosophila, it will be “dme” (Drosophila melanogaster). In this review, as we will mainly refer to human microRNAs, unless stated differently, and we will use the microRNA name without indicating the letter code for species. Orthologous microRNAs in different species will bear the same numbering: for example, hsa-miR-29 in human and mmu-miR-29 in mouse. Lettered suffixes indicate related (paralogous) mature sequences, only differing at one or two positions—for example hsa-miR-29a and hsa-miR-29b, which are expressed from precursors hsa-mir-29a and hsa-mir-29b, respectively. Distinct genomic loci and precursor sequences that express identical mature sequences have numbered suffixes, e.g., hsa-mir-7-1, hsa-mir-7-2, and hsa-mir-7-3. Finally, the mature sequences are assigned names of the form miR-155 (the predominant product) and miR-155* (from the opposite arm of the precursor). A microRNA precursor can give two different mature sequences named -5p (from the 5’ arm) and -3p (from the 3’ arm).
MicroRNAs mediate their regulatory action through binding to the 3’ untranslated region (3’UTR) of target gene mRNAs, usually resulting in translational repression and gene silencing (Bartel et al. 2004 and 2009; Feramisco et al. 2010). MicroRNAs are present in the entire genome, both as intergenic microRNAs and as intronic microRNAs (found in intronic regions of protein-coding transcripts).
RNA interference mediated by microRNAs, is a complex multi-step mechanism including transcription, nuclear processing, nuclear export, and cytoplasmic processing. MicroRNA biogenesis starts from the microRNA gene in the nucleus: intergenic microRNAs are transcribed by polymerase II (Cai et al. 2004; Lee et al. 2004) or polymerase III (Borchert et al. 2006) and give rise to a primary microRNA (pri-miR), whereas intronic microRNAs are co-expressed in the intron region of a protein-coding transcript, through a polymerase II-dependent promoter, and the resulting microRNA precursor (pre-miR) enters the microRNA pathway (Figure 3).
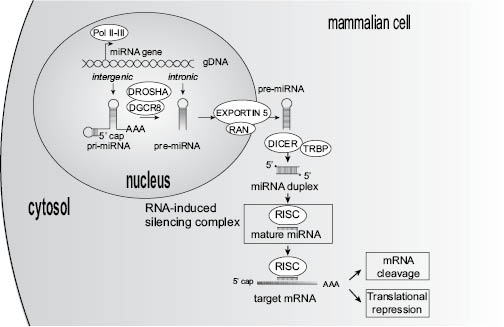
Figure 3: MicroRNAs: nuclear biogenesis, processing, and functions.
Pri-microRNAs are several kilobases long, capped, polyadenylated primary precursors, with hairpin-like structures, possibly polycistronic (i.e., bearing more than one microRNA). The stem-loop structure is cleaved by nuclear RNAse III Drosha, with the help of the double-stranded RNA-binding domain protein DGCR8, to release a microRNA precursor (pre-microRNA) (Cai et al. 2004; Lee et al. 2003; Lee et al. 2011). The Drosha-DGCR8 complex is called the “microprocessor” and allows nuclear maturation of primary microRNAs. All the previous steps occurring in the nucleus, the export of pre-microRNAs to the cytoplasm is mediated via the Exportin-5 protein (XPO5), and its cofactor Ran-GTP (Bohnsack et al. 2004; Lund et al. 2004). In addition to XPO5, Exportin-1 protein (also termed XPO1) can also participate in the export of microRNA precursors through the nuclear membrane, into the cytoplasm (Castanotto et al. 2009). Once in the cytoplasm, pre-microRNAs are further processed by the action of RNAse III DICER1 and TRBP cofactor, which remove the remaining hairpin structures to give birth to a microRNA duplex of 20 to 25 nucleotides in length. Finally, to be capable of modulating gene expression, one of the two strands of the microRNA, the mature microRNA, is loaded into the RNA-Induced Silencing Complex (RISC), which is composed of DICER1, TRBP, and the protein AGO2 (Argonaute 2) (Wilson and Doudna 2013). The integrated microRNA guides RISC to its specific mRNA targets either causing their degradation or directly inhibiting their translation into proteins (Feramisco et al. 2010).
Actually, depending on the degree of complementarity between the microRNA and the target mRNA, two different mechanisms of RISC-mediated gene regulation can occur (Doench et al. 2003; for review Stefani and Slack 2008). When the mature microRNA exhibits total complementarity with the target mRNA, the AGO2 component of RISC is capable of degrading mRNA, whereas if base pairing is incomplete, the silencing is achieved by preventing translation. Hence, microRNAs are capable of decreasing translation of human genes through two different silencing mechanisms. Moreover, each microRNA might target several messenger RNAs (Lim et al. 2005), and a single messenger RNA can also be targeted by multiple microRNAs (Brennecke et al. 2005). This increases the level of complexity in microRNA functions.
4.3.3.3 MICRORNAS REGULATE VARIOUS ASPECTS OF HUMAN PHYSIOLOGY AND EPIGENETICS
It has been estimated that human microRNA repertoire could reach or exceed 2000 genes, and that microRNAs may target 30% to 60% of the transcripts in humans (Friedman et al. 2009). Since microRNAs were recognized as a distinct class of biologic regulators with conserved functions, research has revealed their multiple roles in post-transcriptional regulation of gene expression, through transcript degradation and translational suppression. Different sets of expressed microRNAs are found in different cell types and tissues (Lagos-Quintana et al. 2002). For example, among all microRNAs showing a cell-specific location/expression, miR-203 expression is restricted to keratinocytes (Sonkoly et al. 2007). By affecting gene regulation, microRNAs are likely to be involved in the main biological and physiological processes such as development and organogenesis (Stefani and Slack, 2008; Zhao et al. 2005), differentiation (Chen et al. 2004) proliferation and apoptosis (O’Donnell et al. 2005), growth control, stem cell activity, homeostasis, metabolism, signal transduction, and many others.
All the known mechanisms capable of altering the normal expression of a gene can affect the expression of a microRNA, including mutations and deletions, amplifications, chromosomal translocations, or transcriptional regulation dysfunction, leading to aberrant and dysregulated microRNA expression. Aberrant expression of microRNAs has been observed in numerous disease states, and microRNA-based therapies are under investigations (Tran et al. 2008; Li et al. 2009b; Fasanaro et al. 2010; Zhang et al. 2013).
4.3.3.4 MICRORNAS AND SKIN PHYSIOLOGY
The skin is the largest organ of the body, representing 15% of total body weight in the adult. Human skin shows a highly complex organization and involves the communication of multiple cell types. Skin is composed of three layers: the epidermis, anchored to a basement membrane (the dermo-epidermal junction), delimiting the underlying dermis and finally, subcutaneous hypodermis containing adipocytes. Skin is interspersed with various structures, such as hair follicles, sebaceous glands and sweat glands, blood vessels, lymphatics, and nerves.
Epidermis, the outermost layer of the skin, is a keratinized stratified squamous epithelium providing chemical, physical, and mechanical protection and serving as a first barrier to microbial infection. During the human embryonic development, epidermis is first constituted by a single-cell layer of ectodermal origin, the periderm, that will undergo a stratification process allowing the formation of the different layers of differentiated cells required to fulfill its barrier function (basal, spinous, and granular layers) (Figure 4). The main types of cells that make up the epidermis are keratinocytes, melanocytes, dendritic cells (including Langerhans cells), T-cells, and Merkel cells. The cells of the proliferative basal layer will alternatively differentiate into hair follicles.
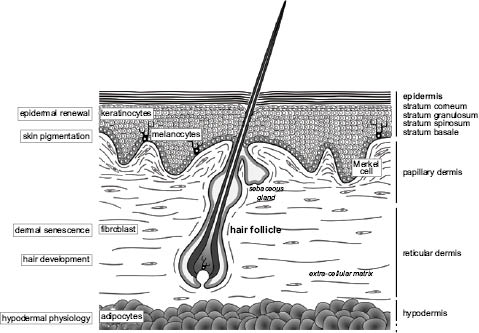
Figure 4: Skin architecture and main processes regulated by microRNAs.
The mesenchymally derived dermis, mainly composed of matrix components (collagens, elastin, fibrillar proteins), also contains also cells, among which are fibroblasts, and immune cells such as mast cells, macrophages, dendritic cells, innate lymphoid cells, and T-cells (Heath and Carbone 2013). The integrity of the dermo-epidermal junction (DEJ) area, located between the epidermis and the dermis, is essential to ensure skin cohesion. Capillary blood and lymphatic vessels are spread throughout the dermis, and nerves access the dermis and the epidermis.
b. MicroRNAs and cutaneous biology
In the skin, microRNA-mediated gene regulation is involved in the main biological processes. To our knowledge, microRNAs were observed and described in skin for the first time in 2005 (Krützfeldt et al. 2005). The evidence of microRNA implication in skin has been demonstrated by the knockdown of DICER1 gene in mouse embryonic skin progenitors, which led to the epidermis-specific removal of microRNA activity and to a disturbed epidermal morphogenesis (Yi et al. 2006).
Many studies revealed that microRNAs are involved in skin homeostasis and cellular processes including development, stem-cell differentiation, signal transduction, metabolism, genomic stability, apoptosis, hair follicle development, skin and hair pigmentation, wound-healing and inflammation (Shilo et al. 2007; Bostjancic and Glavac 2008; Sand et al. 2009; Feramisco et al. 2010; Hildebrand et al. 2010). MicroRNAs are also dysregulated in melanoma and nonmelanoma skin cancer (Kunz 2013; Sand et al. 2012)
Sets of microRNAs were identified as being important in skin physiology and function:
Holst and collaborators conducted a microRNA expression profiling study, in skin biopsies from eight healthy volunteers, and pointed out the 31 most abundantly expressed microRNA species in human skin (Holst et al. 2010). These microRNAs were selected because they had an expression level exceeding 100-fold over the background. MiR-574 appeared to be the most expressed, with almost a 450-fold expression compared to background. The microRNAs identified in this study are as follow (in increasing abundance): miR-203, miR-200c, miR-296-3p, miR-765, miR-539, miR-34a, miR-323-5p, miR-638, miR-608, miR-122, miR-145, miR-23b, miR-768-5p, miR-23a, miR-768-3p, miR-32, miR-206, miR-210, miR-205, miR-297, miR-672, miR-125b, miR-923, miR-574-5p, miR-595, miR-654-5p, let-7f, let-7d, let-7a, let-7c, and let-7b (Holst et al. 2010).
As a review, Aberdam and collaborators set-up a list of 34 microRNAs expressed in the epidermis: miR-15b, miR-16, miR-17, miR-18, miR-19b, miR-20, miR-21, miR-24, miR-27a, miR-27b, miR-30b, miR-34a, miR-92, miR-93, miR-99b, miR-106b, miR-125a, miR-125b, miR-127, miR-130a, miR-133b, miR-141, miR-191, miR-200a, miR-200b, miR-200c, miR-203, miR-205, miR-429, Let-7b, Let-7c, Let-7f, Let-7g, Let-7i (Aberdam et al. 2008).
Studies of the miRNAome in various pathologies such as psoriasis (Sonkoly et al. 2007, Aberdam et al. 2008, for review), systemic scleroderma (Li et al. 2010), atopic dermatitis (Aberdam et al. 2008, for review), keloids (Li et al. 2013) pointed out the deregulation of the expression of various microRNA sets.
All these studies showed the growing importance of microRNAs at different cell type levels in normal and abnormal skin physiology. The role of particular microRNAs of physiological relevance will be discussed in the following paragraphs.
c. Epidermal renewal and skin barrier
Skin integrity requires an efficient epidermis renewal to maintain the skin barrier function throughout life. The constant renewal of the epidermis is supported by progenitor cells mainly located in the basal cell layer, able to proliferate and to asymmetrically divide to engage suprabasal differentiation (Muffer et al. 2008; Pincelli and Marconi, 2010). These progenitors, also called “keratinocyte stem cells” (KSCs) or “somatic stem cells of the epidermis” (SSCEs) reside in a special microenvironment called niche, in the basal epidermis, and constitute less than 1% of the basal population. A delicate equilibrium between stemness and differentiation controls epidermal renewal. Keratinocyte differentiation is a highly coordinated multi-step process regulated by autocrine and paracrine intercellular signaling mechanisms (such as growth factors . . .), and other external stimuli, leading to the expression/repression of genes and microRNAs. SSCEs are also observed in other compartments such as hair follicles and sebaceous glands, corresponding to cellular reservoirs (Beck and Blanpain 2012).
In the epidermis, several microRNAs regulate keratinocyte differentiation, but the most important player in this process is miR-203 (Yi et al. 2008).
d. MiR-203 is a master regulator of epidermal differentiation:
MiR-203 has been identified as one of the highly expressed microRNA in healthy human skin (Holst et al. 2010). Sonkoly and coworkers showed that miR-203 formed a gradient with low expression in the basal cell layer and high expression in the more differentiated suprabasal layers, and that it was expressed more than 100-fold higher in skin compared to other organs in human, and that miR-203 was specific to keratinocytes (Sonkoly et al. 2007). MiR-203 pattern of expression is in accordance with an implication in suprabasal keratinocyte differentiation (Sonkoly et al. 2010).
Interestingly, miR-203 was shown up-regulated in psoriasis, a pathology in which keratinocytes show abnormal states of proliferation and differentiation (Bostjancic et al. 2008), suggesting that the fine homeostasis of this microRNA is necessary to maintain a healthy skin. MiR-203 has also been recently identified as specifically overexpressed during the earliest process, leading to the specification of embryonic stem cells into keratinocytes (Nissan et al. 2011).
MiR-203 is involved in the transition of KSCs to “transit-amplifying” keratinocytes (Aberdam et al. 2008; Beck and Blanpain, 2012). In the adult epidermis, miR-203 is detectable only in the upper layers (Yi et al. 2008). In addition, it has been shown that miR-203 overexpression–induced keratinocyte differentiation and miR-203 inhibition with a specific anti-miR (anti-miR-203) resulted in decreased expression of involucrin (a protein involved in the cornified cell envelope, in the stratum corneum) after calcium treatment (Sonkoly et al. 2010). MiR-203 permits the switch between basal keratinocyte proliferation and suprabasal differentiation, by repressing stemness, through the post-transcriptional repression of target genes expressed in basal KSCs (Yi et al. 2008; Sonkoly et al. 2010).
e. P63, SOCS3, Zfp281, JUN, and ABL1 are the major mir-203 targets in the epidermis:
Several studies have identified miR-203 target genes in skin. The major target described in KSCs is the transcription factor DNp63, the principal p63 isoform, represents a master regulator of the stratification process and is responsible for maintaining the proliferative potential of the epithelial stem cells. By controlling the expression of DNp63, miR-203 is involved in the transition of “KSCs” to transit-amplifying keratinocytes, behaving as the molecular switch for epithelial stratification (Lena et al. 2008; Aberdam et al. 2008).
SOCS3, a negative regulator of the JAK/signal transducer and activator of transcription (STAT) pathway is another potential target, but this is subject to controversy, as contradictory studies exist: while Lena and coworkers showed that despite bio-informatic alignment of miR-203 with SOCS3 3’UTR, the levels of SOCS3 mRNAs were not decreased with the increase of miR-203 levels, when keratinocytes were stimulated to differentiate in vitro (Lena et al. 2008), another study demonstrated the degradation of SOCS3 transcript in a luciferase reporter assay (Wei et al. 2010).
Evidences indicate that miR-203 can be considered as a tumor suppressor, targeting well-known proto-oncogenes such as c-jun (commonly deregulated in a wide range of cancers, including skin tumors), and c-abl (Abelson murine leukaemia viral oncogene homolog 1, or ABL1). Similarly to other miR-203 targets in the skin, c-jun is preferentially expressed in the basal proliferative layer of the epidermis. The suppression of miR-203 in basal cell carcinoma tumors was associated with a marked increase of c-jun expression (Sonkoly et al. 2012). MiR-203 was also shown to control the expression levels of ABL1, a classic oncogene involved in hematopoietic malignancies, where miR-203 is epigenetically silenced by hypermethylation (Bueno et al. 2008). An additional potential miR-203 target expressed in the basal-layer keratinocytes, is represented by the zinc-finger protein 281 (ZNF281) that may contribute to the basal-suprabasal cell transition (Yi et al. 2006; Aberdam et al. 2008).
Finally, some miR-203 minor target genes such as the endothelin A receptor (EDNRA), and the protein phosphatase eyes absent homolog 4 (EYA4), have also been reported in the basal layer of the epidermis (Hildebrand et al. 2010).
f. Other microRNAs important in epidermal renewal
To analyze changes in microRNA expression during keratinocyte differentiation, the expression of 377 microRNAs has been evaluated after calcium-induced differentiation of primary human keratinocytes in vitro, and compared to microRNA expression patterns observed in epidermal stem cells, transient amplifying or terminally differentiated keratinocytes isolated from human skin (Hildebrand et al. 2010). In these experiments, 13 up-regulated and 1 down-regulated (miR-376a) microRNAs were observed after three days of calcium treatment. After seven days of cultivation in the presence of calcium, differential expression reached the numbers of 55 up-regulated and 8 down-regulated microRNAs. The comparison of these expression patterns with the ones obtained with the transient amplifying or terminally differentiated keratinocytes from skin, allowed the identification of a set of microRNAs that may cooperate during skin differentiation (Hildebrand et al. 2010). Indeed, apart from miR-203, nine additional microRNAs were associated with human keratinocyte differentiation in vitro and in vivo: 8 microRNAs up-regulated during differentiation (miR-23b, miR-26a, miR-27b, miR-95, miR-200a, miR-210, miR-224 and miR-328) and one microRNA down-regulated (miR-376a).
A recent study highlighted the role of miR-574-3p and miR-720 in the regulation of p63 expression, via the protein iASSP (Beck and Blanpain 2012, for review). p63 and iASPP are both expressed in the basal epidermal keratinocytes, and iASPP is involved in the regulation of several members of the desmosomal proteins, tight junction and gap junction components, and b1 integrin. The expression of the protein iASSP, under the control of p63, represses miR-574-3p and miR-720, diminishing their impact on the degradation of p63 transcripts, thus forming a positive autoregulatory feedback loop.
MiR-24 is expressed in the suprabasal layers of human epidermis and induces differentiation in vivo by controlling actin adhesion dynamics and cytoskeletal modification during keratinocyte differentiation (Amelio et al. 2012).
Another study reported that miR-200 and miR-205, both highly expressed in normal skin, targeted ZEB1 and ZEB2, the transcriptional repressors of E-cadherin, playing an essential role in maintaining epithelial stability (Banerjee et al. 2013).
Finally, the miR-193b/355a cluster has been very recently shown to have anti-proliferative and anti-migratory properties in human keratinocytes, consistent with potential tumor suppressor functions in the epidermis (Gastaldi et al. 2013).
Skin pigmentation plays a critical role in skin protection at the cellular/DNA level, particularly at the level of the basal keratinocytes allowing epidermal renewal. Melanocytes synthesize melanin in special organelles called melanosomes, which upon maturation are transferred to keratinocytes, and form a supranuclear melanin cap, thus protecting genomic DNA from harmful effects of solar UV irradiation. Several biological processes are implicated in the control of skin pigmentation: melanocyte signaling pathways involved in the transcriptional regulation of melanogenic enzymes, tyrosinase and related enzymes, melanosomal packaging and transfer, autophagic melanin scavenging in keratinocytes, and inflammation (Cichorek et al. 2013). Each of these processes is potentially regulated by post-transcriptional silencing by microRNAs; however, further research will be needed to elucidate the whole mechanism.
There is a great interest in the scientific and medical community to study microRNAs involved in the progression of melanoma, a most dangerous form of skin cancer affecting melanocytes. Kozubek and colleagues determined the microRNA transcriptomic signature distinguishing specimens of melanoma from normal human skin and set up a list of 40 microRNAs. Top-ten microRNAs deregulated in human melanoma was as follows: miR-205, miR-211, miR-15b, miR-26a, miR-451a, miR-203, miR-23b, miR-26b, miR-877, and let-7i-5p (Kozubek et al. 2013). One remarkable member of this list is miR-203 (down-regulated in melanoma), the well-known regulator of keratinocyte differentiation, that evidently also plays a role in melanocyte pigmentation. Hence, the decrease of miR-203 was described to play a pivotal role in melanoma, through reducing melanosome transport and promoting melanogenesis by targeting kif5b, the kinesin superfamily protein 5b, and through negative regulation of the CREB1/MITF/Rab27a pathway (Noguchi et al. 2013).
Albeit the majority of the studies on microRNAs and pigmentation report the modulation of microRNA expression in melanoma cells, several studies on normal human melanocytes NHEM pointed out the potential role of specific microRNAs in human skin pigmentation:
Wu and colleagues described in 2008 the effect of miR-434 targeting tyrosinase in human melanocytes (Wu et al. 2008). A study in Alpaca melanocytes showed the implication of miR-25 as a regulator of pigmentation, targeting MITF expression (Zhu et al. 2010). The increased expression of miR-145, by targeting genes implicated in melanogenesis (Sox9, Mitf, Tyr, Trp1) and genes important for melanosome transport (Myo5a, Rab27a, and Fscn1), induced depigmentation (Dynoodt et al. 2013). In the same study, several microRNAs were identified after forskolin and UV treatment of mouse melanocytes: some of them were up-regulated (miR-130b, miR-182, and miR-9) or down-regulated (miR-125b, miR-139-5p, miR-145, miR-155, miR-193, miR-206, miR-218, miR-221, miR-222, miR-28, miR-335, miR-365, and miR-455), suggesting their implication in the regulation of skin pigmentation (Dynoodt et al. 2013).
In a recent publication, miR-125b was identified as a potent regulator of melanogenesis, as its expression level was inversely related to pigment levels (Kim et al. 2014). Interestingly, the authors demonstrated a modulation of miR-125b expression by methylation of the MIR125B-1 gene promoter.
Surprisingly, microRNAs originating from other cell types seem to control melanocyte pigmentation: indeed, Kim and colleagues recently demonstrated that miR-675 was secreted in exosomes by keratinocytes, and that it could regulate melanocyte pigmentation through a mechanism involving MITF (Kim et al. 2013). This observation enlarges our vision of the mechanisms of action of microRNAs, and brings to our attention the potential role of extracellular microRNA-containing exosome as cell-cell mediators (Hu et al. 2012). Exosomes containing mRNAs and microRNAs are also important in pathological context, as their content is modified in cancers such as melanoma, and their characterization may lead to the identification of useful diagnostic biomarkers (Xiao et al. 2012).
Related posts:
 – THE CELLULAR WATER PRINCIPLE
– THE CELLULAR WATER PRINCIPLE
 – BIOACTIVES GREEN AND SUSTAINABLE INGREDIENTS FROM BIOTRANSFORMATION AND BIOFERMENTATION
– BIOACTIVES GREEN AND SUSTAINABLE INGREDIENTS FROM BIOTRANSFORMATION AND BIOFERMENTATION
 – CHRONOBIOLOGY OF THE SKIN SKIN CIRCADIAN RHYTHM AND CLOCK GENES: A NEW APPROACH TO SLOWING DOWN THE AGING PROCESS
– CHRONOBIOLOGY OF THE SKIN SKIN CIRCADIAN RHYTHM AND CLOCK GENES: A NEW APPROACH TO SLOWING DOWN THE AGING PROCESS
 – GLYCATION, PROTEASOME ACTIVATION, AND TELOMERE MAINTENANCE
– GLYCATION, PROTEASOME ACTIVATION, AND TELOMERE MAINTENANCE
 – SKIN WHITENER INGREDIENTS
– SKIN WHITENER INGREDIENTS
 – AMINO ACIDS
– AMINO ACIDS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


