Fig. 17.1
12R-LOX and eLOX-3 act in sequence to form R-enantiomeric epoxyalcohols. The dioxygenase 12R-LOX converts PUFA containing a cis-cis-pentadiene structure to the corresponding R-hydroperoxide which in turn is converted by the hydoperoxide isomerase activity of eLOX-3 to the corresponding R-epoxyalcohol derivative. The prototypical LOX substrate arachidonic acid is metabolized via 12R-HPETE to a R-epoxyalcohol of the hepoxylin A-type
Loss-of-Function Mutations in the Genes of 12R-LOX and eLOX-3 are the Second Most Common Cause of ARCI
The discovery of a genetic connection between 12R-LOX and eLOX-3 and an inherited skin disorder points to an essential function of these LOX in the proper functioning of the epidermal barrier. In fact, a major breakthrough in understanding the physiological function of these LOX in skin was provided by a genetic study of Fischer and colleagues in 2002 (Jobard et al. 2002). They showed that mutations in ALOX12B and ALOXE3, the genes encoding 12R-LOX and eLOX-3, are linked to the development of autosomal recessive congenital ichthyosis (ARCI) supporting the long suspected role of LOX in the maintenance of skin permeability barrier.
ARCI refers to nonsyndromic congenital ichthyoses including harlequin ichthyosis, lamellar ichthyosis, and congenital ichthyosiform erythroderma (Oji et al. 2010). This clinically and genetically heterogeneous group of cornification disorders is characterized by a generalized scaling of the skin and has a prevalence of approximately 1 in 200,000 persons in the European and northern American populations (Oji and Traupe 2006). Affected newborns are often encased in a collodion membrane. After loss of this encasement in the first weeks of life, patients exhibit a generalized scaling, which varies in extent, color, and degree of adherence. An underlying mostly mild erythema is often seen. The typical histological phenotype of skin from ichthyosis patients is characterized by variable epidermal hyperplasia and marked hyperkeratosis. It is generally acknowledged that the ARCI phenotype results from a physical compensation for the defective permeability barrier that underlies all ichthyosis disorders.
To date, apart from the two LOX genes, seven other genes have been identified that underlie the ARCI phenotype, including TGM1 encoding transglutaminase 1, ABCA12 encoding a lipid transporter, a cytochrome P450 gene CYP4F22, a putative receptor gene NIPAL4/ ichthyin, two lipase genes LIPN and PNPLA1 and CERS3 encoding ceramide synthase 3 (Eckl et al. 2013; Fischer 2009; Grall et al. 2012; Israeli et al. 2011). Genetic studies indicated that mutations in ALOX12B and ALOXE3 are the second most common cause of ARCI found in about 10 % of the cases (Eckl et al. 2005, 2009; Vahlquist et al. 2010). To the present, 40 mutations have been identified in ALOX12B and 13 in ALOXE3 (Akiyama et al. 2010; Eckl et al. 2005, 2009; Harting et al. 2008; Israeli et al. 2013; Jobard et al. 2002; Lesueur et al. 2007; Rodriguez-Pazos et al. 2011; Vahlquist et al. 2010). Most of the mutations are single nucleotide exchanges and only few are made of small deletions or small inserts. About a quarter of them are nonsense mutations resulting in a premature stop codon while the majority represent missense mutations. Examination of the molecular data revealed allelic heterogeneity for ALOX12B, with only some mutations located in the N-terminal beta barrel domain but most distributed within the catalytic domain with a minor focus on exons 9 and 12. In ALOXE3 two recurrent mutations (p.Arg234X found in 13 and p.Pro630Leu found in 15 alleles) have been identified as mutational hotspots (Eckl et al. 2009; Fischer 2009).
While the nonsense mutations identified in the LOX genes definitely eliminate the catalytic activities the functional impact of the missense mutations on enzyme activities has been evaluated in detail. Although most of the mutations apparently did not interfere with substrate binding or catalysis directly, all of them led to complete loss of the catalytic activity of the LOX enzymes (Eckl et al. 2005, 2009; Yu et al. 2005) supporting the concept that the loss of function of 12R-LOX or eLOX-3 is fundamental to the pathogenesis of the LOX-dependent form of ARCI.
Animal Models to Study LOX Function in Skin
To investigate the physiological role of the 12R-LOX/eLOX-3 pathway and the molecular mechanisms of its action in the formation and maintenance of the epidermal barrier mutant mice for targeted inactivation of the 12R-LOX and eLOX-3 genes were established in our group.
General 12R-LOX and eLOX-3 Gene Knockout Results in Perinatal Death due to Severe Barrier Defects
A conditional null allele of the 12R-LOX gene Alox12b was generated by targeting exon 8 for Cre-mediated deletion and mice with general 12R-LOX gene knockout were obtained by mating mice harboring this mutant allele with transgenic CMV-Cre deleter mice (Fig. 17.2) (Epp et al. 2007). Exon 8 encodes a highly conserved region with two of the iron-binding histidines that are absolutely required for catalytic activity. Thus, deletion was expected to produce a truncated non-functional protein but actually resulted in the complete loss of 12R-LOX protein expression.
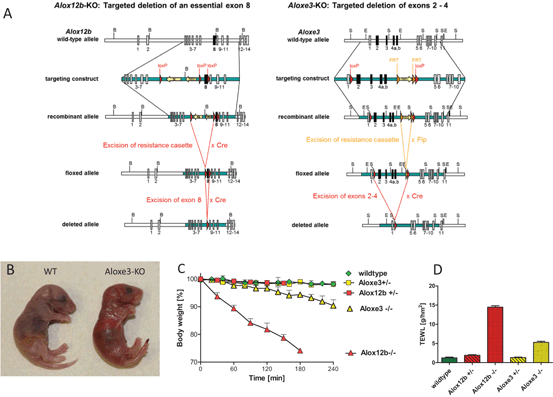
Fig. 17.2
12R-LOX and eLOX-3 knockout models. Targeted disruption of Alox12b and Aloxe3, the genes encoding 12R-LOX and eLOX-3, results in early neonatal death due to permeability barrier defects. a Strategy of LOX gene targeting using Cre-LoxP and Flp-FRT technology for the generation of inactivated LOX alleles. b Cross morphology of eLOX-3 deficient neonates. Homozygous knockout mice are born with red, shiny skin that rapidly desiccates. c Dehydration over time as indicated by loss of body weight. d Transepidermal water loss (TEWL) is increased up to eightfold in 12R-LOX deficient mice and about four-fold in eLOX-3 knockouts
While heterozygous mice with disrupted Alox12b allele did not show any phenotype and reproduced normally homozygous mutant mice died within few hours after birth due to severely impaired barrier function. The mutant mice were born with red, shiny skin that rapidly desiccated. The neonates did not feed and lost 10 % of their weight per hour owing to an eight-fold increased transepidermal water loss (TEWL) as compared to wild-type and heterozygous littermates. Dye penetration assays indicated that both the inside-out and the outside-in water barrier function were severely affected. The same lethal phenotype associated with 12R-LOX deficiency was reported by another group for an ethylnitrosurea-induced mouse mutant harboring a loss-of function mutation in Alox12b (Moran et al. 2007).
With the exception of a more tightly packed stratum corneum the neonatal 12R-LOX-deficient skin did not show gross abnormalities in the structural organisation of the epidermis (Epp et al. 2007). Only at the ultrastructural level, anomalies were observed including vesicular structures in the upper granular layers of the epidermis pointing to perturbance of the assembly/extrusion of lamellar bodies (LB) that play a crucial role in formation of the water impermeable barrier. Studies on the molecular mechanisms involved in the disruption of the barrier function showed that 12R-LOX-deficiency severely affected lipid components of the barrier but also proteins of the CE. Complete lack of filaggrin monomers, crucial components of the CE, associated with enhanced levels of high molecular intermediate species indicated impairment of the proteolytic processing of profilaggrin to filaggrin in 12R-LOX knockout epidermis. The loss of monomeric filaggrin was associated with increased fragility of mutant corneocytes supposedly contributing to the early neonatal death of the mutant mice. Moreover, a disordered composition of ceramides, in particular a decrease of protein-bound ceramide species could be identified as the decisive defect compromising barrier function in 12R-LOX-deficient skin. Ester-bound ceramides are the major constituents of the CLE. The CLE is formed by transglutaminase 1-catalyzed covalent attachment of the ceramides ω-hydroxyacyl-sphingosine (OS) and their sphingosine-free derivatives to involucrin and other CE peptides. The OS species especially those containing very long chain fatty acids (VLC-FA > C32) were severely reduced to only a few percent of the wild-type level. In line with these findings, Brash and coworkers showed that the CLE was largely absent in the epidermis of 12R-LOX deficient mice (Zheng et al. 2011).
For inactivating the eLOX-3 gene Aloxe3 the Cre-loxP and flp-FRT systems were used to generate a conditional null allele with two loxP sites flanking exons 2–5 (Krieg et al. 2013). Cre-mediated deletion of the floxed segment results in an out of frame-fusion of the remaining exons thus causing a premature stop codon in exon 6 (Fig. 17.2). In the constitutive knockout model, homozygous mutant mice developed a similar but somewhat less severe skin phenotype as observed in the 12R-LOX knockout animals. Whereas 12R-LOX knockout mice all died rapidly within 3 h after birth, the eLOX-3 knockouts survived up to 12 h exhibiting less severe water loss as indicated by four-fold elevated TEWL. Accordingly, eLOX-3-deficient skin showed less pronounced structural and biochemical abnormalities, notably lack of abnormal vesicular structures and unimpaired profilaggrin processing. Of importance, lipid analysis demonstrated that the severity of barrier failure was related to the level of reduction in covalently bound ceramides, which was in eLOX-3 knockout skin to about half of the normal level as compared to only a few percent in 12R-LOX-deficient mice.
Strongly reduced levels of hepoxilin metabolites in eLOX-3-deficient epidermis furthermore indicated a function of eLOX-3 in mammalian skin as hepoxilin synthase linked to the 12S-LOX pathway. Hepoxilins and their triol derivatives have been found in normal human skin and in elevated levels in human psoriatic scales (Anton et al. 1998; Anton and Vila 2000). These metabolites have been shown to play a role in a variety of physiological processes including inflammation and neurotransmission. The majority of activities appear to be mediated by the stimulation of intracellular calcium or increased calcium transport across the membrane (Pace-Asciak 2009). In skin various potential mechanisms of action for these metabolites have been discussed including an action via a putative membrane receptor NIPAL/ichthyin or through activation of PPAR receptors (Lefevre et al. 2004; Yu et al. 2007). The exact mode of action and physiological role of hepoxilin-type metabolites in epidermis, however, remains to be established.
Adult 12R-LOX-Deficient Skin Develops an Ichthyosiform Phenotype
As a compensatory response to the excessive water loss impaired barrier function typically results in the development of a skin phenotype characterized by thickening of the epidermis (epidermal hyperplasia) and abnormal formation and desquamation of the SC (hyperkeratosis). Although some morphological and biochemical anomalies indicated the onset of hyperkeratosis and defects in or delay of the desquamation process an obvious cutaneous phenotype was not observed in the neonatal skin of LOX-deficient mice, supposedly due to their early neonatal death. By utilizing neonatal skin grafting and the conditional knockout model with inducible gene inactivation in the skin the adult skin phenotype of 12R-LOX-deficient skin could be analyzed. Alox12b-deficient skin that matured after transplantation developed an ichthyosiform appearance with thickening of the epidermis, hyperproliferation, hypergranulosis, parakeratosis, and severe hyperkeratosis recapitulating the phenotype observed in skin from ichthyosis patients (De Juanes et al. 2009). Biochemically, the grafted skin showed restoration of profilaggrin processing and overexpression of CE components such as filaggrin, involucrin, and repetin. As a result, the TEWL was remarkably decreased as compared to neonatal knockout skin but still higher as compared to normal skin indicating improvement of the barrier defect by compensatory hyperkeratosis.
For inducible LOX ablation in the skin mice harboring the floxed LOX gene alleles were mated with a transgenic mouse line expressing a tamoxifen-dependent Cre recombinase under the control of the Keratin 14 promoter. Tamoxifen induced ablation of 12R-LOX, and similarly of eLOX-3, resulted in the development of a severe phenotype associated with growth retardation, dramatic loss of body weight and premature death (unpublished data). Morphological changes observed in mutant mice included focal alopecia, scaling of the skin and palmoplantar keratoderma while histological analyses showed hyperplasia and strong hyperkeratosis resembling the ichthyosis phenotype.
Organotypic Mouse Skin Equivalents
With the aim to obtain an experimental in vitro model to study molecular mechanisms of LOX action more closely organotypic mouse skin models were established. (Rosenberger et al. 2014). These in vitro systems are three-dimensional two-layered configurations consisting of epidermal keratinocytes growing on top of a dermal equivalent. The keratinocytes are freshly isolated from skins of neonatal mice and cocultivated with dermal mouse fibroblasts which are embedded in a scaffold of native collagen type I obtained from rat tail tendon. Closely resembling the in vivo situation these skin equivalents grow air-exposed with nutrients being supplied from underneath by diffusion of medium of a defined composition.
With this setup the keratinocytes are able to undergo proper epidermal morphogenesis. They form a multilayered well organized stratified epithelium consisting of basal, spinous, granular and cornified strata closely resembling the natural tissue architecture found in native mouse skin. The keratinocytes of the different layers of the epidermal equivalent show the respective characteristic morphological features and properly express various differentiation markers, such as keratin 1 and 10, filaggrin, involucrin, loricrin, and repetin (Fig. 17.3).
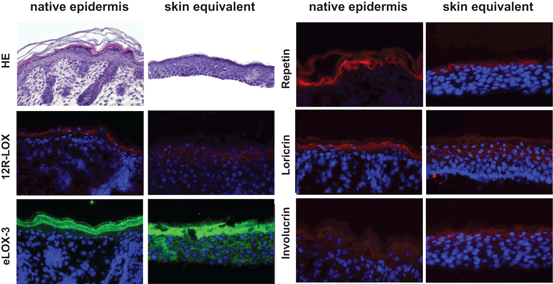
Fig. 17.3
Organotypic mouse skin equivalents. Skin equivalents established from primary keratinocytes represent suitable in vitro models resembling the natural tissue architecture and expression pattern of LOX and epidermal differentiation markers as found in native mouse skin
The major components of tight junctions are present as well as epidermal barrier lipids. Furthermore ultrastructural analyses indicate that these components are not only present but that they are processed and assembled correctly forming all structures of the epidermal barrier. These results indicate that our organotypic mouse skin models are valuable tools to study the molecular basis for epidermal barrier defects due to LOX deficiency and to test potential therapeutic approaches.
Our Current View on the Mechanism of Action of 12R-LOX/eLOX-3 in Barrier Function: Induction of Structural Changes Rather than Eicosanoid Signaling
Spatial and temporal colocalisation and a differentiation-dependent expression pattern in epithelial tissues early on suggested a common physiological role of 12R-LOX and eLOX-3 (Krieg et al. 2001). Their pivotal role in epidermal barrier function was unveiled by the finding that inactivating mutations in the genes of 12R-LOX and eLOX-3 are linked to the development of ARCI (Jobard et al. 2002). The fact that the same skin phenotype is caused by mutation of either gene suggested that both LOX may function in the same metabolic pathway and that the product of one of these enzymes may be the substrate of the other. Biochemical studies indeed showed that the two enzymes act in tandem to convert fatty acid substrates via R-hydroperoxides to specific epoxyalcohol derivatives (Yu et al. 2003). Thus, the common LOX substrate arachidonic acid is metabolized via 12R-HPETE to a R-epoxyalcohol of the hepoxilin family (Fig. 17.1). As 12R-HPETE turned out to be a preferred substrate of eLOX-3 in vitro it appeared that this specific hepoxilin pathway is the mechanistic link between 12R-LOX/eLOX-3 and their action in barrier function (Yu et al. 2006).
This view, however, is compromised by the following facts: (i) In contrast to the human enzyme the mouse 12R-LOX does not metabolize free arachidonic acid but only esterified substrates raising the question as to the functional homology of the mouse and human enzyme and to the nature of the natural substrate for this enzyme (Siebert et al. 2001). (ii) The data from our 12R-LOX-deficient mouse models clearly documented a functional homology of the mouse and human enzymes in skin. (iii) Several lines of evidence from fatty acid analyses and from feeding studies in animals indicate that not arachidonic acid but linoleic acid is the physiologically relevant EFA in relation to mammalian epidermal barrier function. (iv) Our studies furthermore revealed that the loss of covalently bound ceramide species as the major constituents of the CLE may be the decisive defect compromising barrier function in LOX-deficient skin.
Putting together these findings Brash and colleagues then recently suggested a new model of the action of LOX in barrier function that links the 12R-LOX/eLOX-3 pathway to oxidation of linoleate-containing ceramides and consequently to the formation of CLE (Zheng et al. 2011). The CLE is a hydrophobic envelope surrounding the corneocytes that is assumed to play an important role as a scaffold for the extracellular lipid lamellae or in corneocyte cohesion (Elias et al. 2000). This structure is enriched in ω-hydroxy ceramides (ω-hydroxyacyl-sphingosine, OS) and very long chain fatty acids (VLC-FA) linked covalently by ester bonds to glutamine/glutamate residues within the outer CE. The CLE is presumed to originate from LB by fusion of their limiting membrane with the apical plasma membrane of the outermost stratum granulosum cells (Fig. 17.4). The LB membrane is enriched in glycosylated linoleate-containing esterified ω-hydroxyacyl-sphingosine ceramides (glucosyl-EOS), the precursor molecules of the CLE constituents. Upon fusion and secretion a multistep processing of glucosyl-EOS has been proposed to occur including deglucosylation by glucocerebrosidase, hydrolysis of the linoleate to form ω-hydroxy ceramide and covalent attachment to the CE by transglutaminase. The fatty acid moiety of the CLE originates from ceramidase-catalyzed hydrolysis of OS which may occur before or after the attachment step.
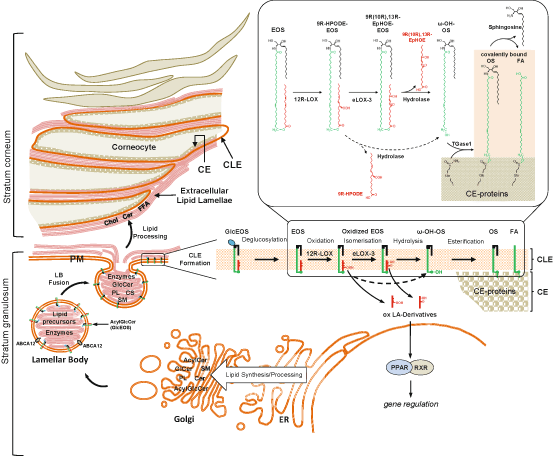
Fig. 17.4




Schema illustrating the formation of lipid structures essential for the epidermal barrier function and the role of 12R-LOX/eLOX-3 in these processes. The permeability barrier of the skin is localized in the stratum corneum. This function mainly resides in the extracellular lipid domains (extracellular lipid lamellae) between the outermost enucleated cells (corneocytes). The corneocyte is surrounded by an inner protein envelope (CE) and an outer corneocyte lipid envelope (CLE), a lipid monolayer covalently bound to the CE. The CLE is thought to be indispensable as a scaffold for the lamellar organization of the extracellular lipids. The lipid components of these structures originate from precursor lipids synthesized and processed mainly in the endoplasmic reticulum (ER) and Golgi apparatus. The precursor lipids, including acylceramide (AcylCer), glucosylceramide (GlcCer), sphingomyelin (SM), glycerophospholipids (PL), and cholesterol sulfate (CS), are packaged together with processing enzymes into lamellar bodies (LB) within the upper cell layers of the epidermis. Lipid packaging into the LB requires function of ABC transporter protein, ABCA12, localized to the limiting membranes of the LB. This LB membrane furthermore, is enriched in acylglucosylceramides (AcylGlcCer, including GlcEOS), the precursor molecules of the CLE constituents. Fusion of the LB with the apical plasma membrane (PM) in the uppermost nucleated cell layer of the epidermis (stratum granulosum), allows extrusion of lipid precursors into the extracellular domains. Subsequent enzymatic processing of precursor lipids generates the major lipid classes forming the extracellular lipid lamellae, i.e. ceramides (Cer), cholesterol (Chol), and free fatty acids (FFA). Formation of the CLE requires several processing steps of acyl- GlcCer including an essential 12R-LOX/eLOX-3 catalysed step. The successive oxygenation of the linoleate moiety of EOS by 12R-LOX and eLOX-3 generates an “oxygen signal” that permits esterase-catalyzed hydrolysis of the oxidized linoleate to release the ω-hydroxyl function on the ceramides. The ω-OH group of these Cer is required for CLE formation, as this moiety represents the attachment point between the Cer and specific amino acids of cornified cell envelope (CE) to be crosslinked by transglutaminase 1. The released oxidized linoleic acid derivatives (ox LA-Derivatives), 9R-HPODE and the hepoxilin-like metabolite (9R(10R), 13R-EpHOE), are hypothesized to act as ligands for nuclear hormone receptors (PPAR/RXR) that regulate genes involved in epidermal differentiation
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
