Fig. 2.1
Pathologic staging of melanoma progression and 5-year survival rates. The later stage of melanoma at the time of diagnosis is directly related to 5-year survival rates, as adapted from the AJCC melanoma staging database. Pathologic staging includes 1A–1B, 2A–2C, 3A–C, and stage 4. Breslow thickness, which is measured in millimeters from the stratum granulosum of the epidermis, also correlates with disease progression and survival
A number of molecular alterations have also been associated with the progression of melanoma. Early in the development of cancer, cells obtain the ability to undergo uncontrolled proliferation. Two pathways that are known to regulate cell proliferation are the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K) pathways, and both have been found to be deregulated early in melanoma formation (Fig. 2.2). B-RAF, a serine-threonine kinase downstream of RAS in the MAPK pathway, is mutated in 80 % of nevi [91]. This mutation produces a constitutively active B-RAF, resulting in increased proliferation. Additionally, cell cycle arrest that may occur in response to oncogenic B-RAF activity is impaired by secondary inactivation of the CDKN2A locus, discussed in detail below. PI3K pathway activity is increased in melanoma progression by loss of its negative regulator, the tumor suppressor phosphatase and tensin homolog (PTEN). Cell proliferation is furthermore enhanced by increased expression and activity of cell cycle-regulated proteins, such as cyclin D. These highly proliferative cells then become malignant following enhanced cell motility and invasion through alteration of protein expression. These modifications include loss of E-cadherin, increased N-cadherin, as well as increased matrix metalloproteinase 2 (MMP-2) expression [83].
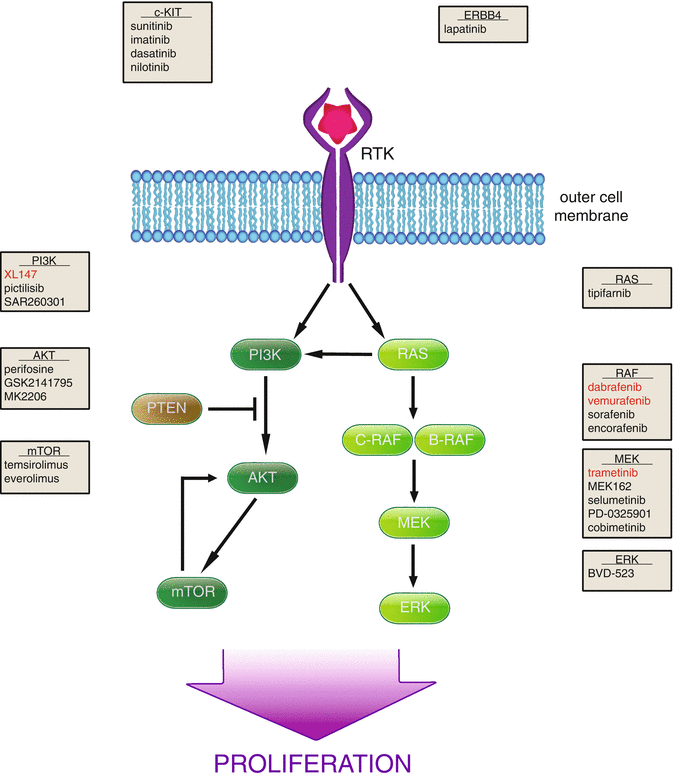
Fig. 2.2
MAPK and PI3K pathway targeting in melanoma. Upon ligand binding to transmembrane receptors, survival pathways such as PI3K (left) and MAPK (right) are stimulated, resulting in increased cell proliferation. Multiple members of these pathways are being explored as targets in therapy, and the protein targeted as well as the corresponding inhibitors are given in gray boxes. Red writing indicates FDA approval in melanoma
Not only are certain acquired mutations associated with the disease progression, but also inherited mutations can enhance the likelihood of developing melanoma. Ten percent of melanoma patients have a documented family history of melanoma [42]. The most common genetic alteration found in familial melanoma is that of CDKN2A. It is estimated that around 40 % of familial melanoma subjects carry a CDKN2A alteration [44, 81]. Thirty percent of CDKN2A mutation carriers develop melanoma by age 30 [11]. CDKN2A encodes two regulators of the cell cycle, p16INK4a and p14ARF. It is through inactivation of this locus that cancer cells are able to evade arrest and enhance proliferation by two mechanisms (Fig. 2.3) [123]. Normally p14ARF is able to inhibit the function of the ubiquitin ligase for p53, HDM2, resulting in increased p53 levels and cell cycle arrest. Without proper p14ARF function, HDM2 is free to target p53 for degradation, bypassing arrest mediated by p53. In addition to p14ARF, CDKN2A encodes p16INK4a. p16INK4a normally halts cell division at the G1-S checkpoint by inhibiting cyclin-dependent kinase 4 (CDK4), preventing Rb phosphorylation. Without this protein, Rb is phosphorylated and cancer cells are able to move into S phase, effectively evading another cell cycle checkpoint.
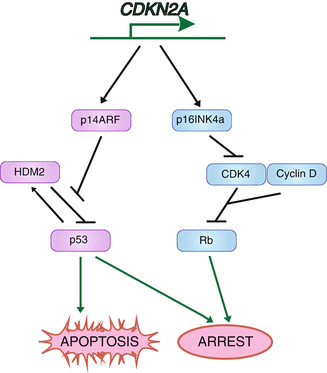
Fig. 2.3
Protein products and signaling pathways of the CDKN2A gene. The gene CDKN2A encodes p14ARF as well as p16INK4a, which are both involved in cell cycle control and can result in apoptosis or arrest
Germline mutation of the CDK4 gene itself is also found in melanoma-prone families [103]. CDK4 binds cyclin D and promotes cell cycle progression. Mutation of CDK4, occurring at arginine 24, renders CDK4 unable to bind its inhibitor p16INK4a, resulting in increased activity [125]. Both cyclin D overexpression and CDK4 mutation are found in melanoma, enhancing cell proliferation by the same network.
Microphthalmia-associated transcription factor (MITF) is another gene linked to familial melanomas and is amplified in 15 % of cases [124]. MITF is known as the master regulator of melanocyte differentiation, and mutation and loss of function of MITF in mice cause complete absence of the melanocyte lineage [70]. Increased in melanoma, MITF has been found to regulate multiple target genes important for cellular survival and proliferation [70]. Surprisingly, MITF expression has also been shown to inhibit melanoma cell proliferation [14, 71]. These conflicting results are reconciled by the rheostat model, where either extreme of expression (i.e., very high or very low) results in arrest or apoptosis, while intermediate levels enhance proliferation [46].
In addition to proteins that regulate the cell cycle, there are other altered cellular mechanisms known to enhance melanoma susceptibility. UV light, mainly UV-B, has specifically been linked to melanoma acquisition, as UV radiation is able to induce DNA damage. Melanocytes have developed a mechanism to produce the pigment melanin, which can shield DNA from incoming UV rays. The lack of skin pigment is one reason melanoma occurs far more frequently in those with lighter skin who sunburn easily [60]. Additionally, dysfunction in the regulation of the melanin production pathway is connected to familial melanoma. Melanocortin 1 receptor (MC1R) is important for UV-induced skin pigmentation in response to its ligand, α-melanocyte-stimulating hormone (α-MSH) [7]. Without proper MC1R function, the ability of melanocytes to produce melanin (specifically eumelanin) is impaired, leaving DNA exposed to incoming UV irradiation, therefore increasing susceptibility to melanoma [111]. It is important to note that non-UV-induced melanomas also occur. In mice that lack MC1R activity (and therefore eumelanin production) and harbor mutant B-RAF, invasive melanomas arose without any UV exposure. This was found to be due to increased pheomelanin synthesis and resulting oxidative DNA damage [86].
Surprisingly, the mutations common in cutaneous melanoma, such as B–RAF and N–RAS, are not found in uveal melanoma [20, 95]. Uveal melanoma, which arises from melanocytes in the choroidal plexus of the eye, comprises 5 % of diagnosed melanomas and portrays a distinct landscape of mutations. For example, BRCA1-associated protein 1 (BAP1) is mutated and activity lost in 84 % of metastatic uveal melanomas [53]. While germline mutation of BAP1 results in cancer predisposition, it also is found in spontaneous melanomas, with the highest prevalence in uveal melanoma (40 %) [117]. Another mutation also appears highly specific for uveal melanoma, GNAQ. GNAQ, a G-protein α-subunit, has an activating mutation in 46 % of uveal melanoma [112]. Even though all melanomas arise from melanocytes, not all present the same tendencies for mutations, highlighting the necessity for individualized molecular profiling before treatment.
One of the oddities of melanoma is the low prevalence of TP53 mutations. p53, encoded by the gene TP53, is a tumor suppressor protein that in response to cell stress and DNA damage is increased and can result in cell cycle arrest or apoptosis [15]. While p53 is mutated in about half of cancers, mutation frequency in melanoma is very low, around 9 % [48]. Initial studies found high levels of p53 in many melanoma samples when staining by immunohistochemistry, which is normally indicative of accumulated mutant p53. However, these results turned out to be misleading. In fact, not only do melanomas rarely mutate TP53, but they are also found to express high levels of wild-type (wt) p53 [3]. It remains unclear what function high levels of wt p53 play in melanoma, although it is proposed that p53 activity is modified. Indeed, other members of the p53 pathway are found to be deregulated in melanoma. This includes loss of p14ARF and also amplification of HDM2, leading to increased inhibition and degradation of p53. Other proteins have also been found to alter the transcriptional ability of p53. Recent reports have attributed elevated levels of iASPP in modulating p53 transcriptional function [73]. While p53 is not commonly mutated in melanoma, its levels and activity are modulated, leading to a functional impairment.
Multiple acquired and inherited mutations are known to be prevalent in melanoma. These include proteins involved in different aspects of cancer progression, from growth and proliferation to loss of cell adhesion and increased invasion. Many of these proteins are being actively pursued for treatment therapies and will be discussed in detail below.
2.4 B-RAF Mutation and Targeted Therapy
B-RAF mutations are found in about 50 % of melanomas, and 90 % of these mutations occur at amino acid position 600. Furthermore, the vast majority of these mutations substitute the amino acid valine to glutamic acid (V600E) [24]. Not only is mutant B-RAF common in melanoma, it is also linked to more aggressive disease. While time of metastasis appearance from initial diagnosis was not affected by the presence of wt vs. mutant B-RAF, overall survival was shortened in patients that harbored mutant B-RAF from 8.5 to 5.7 months [72]. However, no prognostic impact of BRAFV600 mutations on overall survival was observed for patients with primary melanoma and also not for patients with distant metastasis treated with monochemotherapy [79, 80]. B-RAF is activated by RAS, as is its family member C-RAF, and both are able to activate MEK, the next kinase in the MAPK cascade (Fig. 2.2). Additionally, in N-RAS mutant melanoma C-RAF is preferentially activated over B-RAF [28]. Interestingly, unlike B-RAF, there are no known mutations of C-RAF in melanoma [32].
The first small molecule inhibitor to be tested in melanoma patients was sorafenib, a broad-spectrum tyrosine and serine-threonine kinase inhibitor. While sorafenib showed activity against B-RAF, it also inhibits C-RAF and other kinases such as PDGFR, VEGFR-2, and c-KIT [118]. Unfortunately, in clinical trials no benefit was found in those treated with sorafenib [30]. This may be due to the fact that even at the maximum tolerated dose, B-RAF was not inhibited sufficiently. This could be attributed to the inhibition of other kinases, resulting in counteractive effects and/or increased toxicity. In efforts to achieve more robust B-RAF inhibition, selective B-RAF inhibitors, such as PLX4720 and PLX4032 (vemurafenib), were generated [68, 110].
Vemurafenib and dabrafenib both selectively inhibit B-RAF, including the V600 mutant. In clinical trials, 85 % of patients saw some tumor regression, an unprecedented robust response. Unfortunately, it was shortly discovered that the median response for progression-free survival was only 5–7 months [107]. Interestingly, use of B-RAF inhibitors has shown to result in spontaneous growth of squamous cell carcinomas and keratoacanthomas in 20 % of patients. This is partly due to developed RAS mutation, which preferentially signals through C-RAF, not B-RAF. Fortunately, these lesions are easily removed by surgical resection [106]. Additionally, it has been discovered that B-RAF inhibition in B-RAF mutant melanoma cell lines results in loss of signaling through pERK, but in activation of the MAPK pathway in wt B-RAF lines. This is due to inhibitor binding to B-RAF increasing hetero- and homo-dimerization of B-RAF and C-RAF, leading to activation of C-RAF and increased downstream signaling [54]. These data highlight the necessity of knowing the molecular profile of tumors before therapy is designed. Not only do these small molecules have a low toxicity, they also had a high response rate. Unfortunately, relapse was common as the disease was able to compensate for B-RAF inhibition. These results, however, were very promising for future development of small molecule inhibitors.
In an effort to treat patients who have relapsed after B-RAF inhibition, multiple studies were undertaken to determine the mechanism of compensation within tumor cells. B-RAF itself was probed for second mutations that would hinder small molecule inhibitor binding, but none were found [113]. However, alternative splicing of B-RAF has been seen, resulting in a protein that can still bind the inhibitor but no longer binds RAS due to a deletion in that region, resulting in increased dimer formation and activity [92].
In addition to B-RAF splicing, it was more commonly observed that the MAPK pathway was reactivated by other means. This either occurred upstream by mutation of RAS or upregulation of tyrosine kinase receptors such as PDGFR and ERBB2, as well as upregulation of C-RAF [34]. Downstream members of the pathway were also affected, as activating mutations of MEK, or amplification of the gene encoding COT, were observed [61, 115]. Outside of the MAPK pathway, resistance also occurred through increased activity of a parallel pro-proliferative pathway, PI3K. All of these mediators of resistance exhibited by melanoma are now possible therapeutic targets.
2.5 The Role of N-RAS in Melanoma
N-RAS is found to be mutated in 15–30 % of melanomas. Interestingly, N-RAS mutations rarely overlap with B-RAF mutation, likely due to the redundancy of pathway activation this could create [24, 43]. The common mutations of N-RAS are substitutions at position Q61 (around 86 %) and result in an inability of N-RAS to hydrolyze GTP to GDP, resulting in a constitutively active kinase [36]. N-RAS activity can also be upregulated in melanoma by increased levels of its upstream RTKs, such as c-KIT, c-MET, and EGFR [6, 40].
Tipifarnib (R115777) is a farnesyltransferase inhibitor (FTI) currently being tested in melanoma to inhibit RAS activity. Farnesylation is an important posttranslational modification of RAS that promotes its localization to the cell membrane, which is necessary for activation. Treatment with tipifarnib did result in decreased RAS activity, as indicated by the loss of AKT and ERK phosphorylation, in tumor samples taken from patients. Unfortunately, there was no clinical response observed [41]. These results show that farnesyltransferase inhibition alone does not cause tumor regression in advanced melanoma, and a more specific RAS drug may be more efficacious. Additionally, downstream components of pathways mediated by RAS (MAPK and PI3K) are also attractive targets for melanoma, and many have multiple small molecule inhibitors already being explored.
2.6 MAPK Pathway Inhibition
Downstream of RAF in the MAPK pathway are MEK and ERK, both of which are under investigation as targets in melanoma therapy. Currently, there is one inhibitor specific for MEK1 and MEK2 which is FDA approved, trametinib. In a phase 3 clinical trial, trametinib was compared to standard dacarbazine treatment in patients with metastatic melanoma harboring mutant B-RAF. Those given trametinib had better progression-free survival and increased overall survival when compared to the chemotherapy group, with 81 % survival at 6 months compared to 67 %, respectively [39].
In addition to trametinib, multiple other MEK inhibitors are being explored in melanoma. These include MEK162, which has been tested in both B-RAF mutant and N-RAS mutant (B-RAF wild-type) patients, and both sets achieved 20 % partial response at 3.3 months [4]. However, there is dose-limiting toxicity because the MAPK pathway is blocked in all cells, not just those that are cancerous.
Selumetinib (AZD6244), a MEK1/2 inhibitor, was shown to suppress melanoma tumor growth in mice, and tumor regression was enhanced with combination with docetaxel, compared to either treatment alone [50]. This combination therapy is now currently undergoing clinical trial (NCT01256359). Other trials have also tested selumetinib for efficacy in patients with unresectable advanced melanoma. When compared to temozolomide treatment, selumetinib did not have a significant effect on progression-free survival [2]. However, of the partial responders to selumetinib, 83 % were B-RAF mutant [65]. These data led to the possibility that this MEK inhibitor would be most effective in patients with mutant B-RAF. Later, a clinical trial compared selumetinib in combination with dacarbazine vs. placebo with dacarbazine in mutant B-RAF patients with advanced melanoma. Progression-free survival was slightly improved with those treated with selumetinib in combination with dacarbazine, but unfortunately no significant difference in overall survival was observed [96].
As MEK inhibition proved to be effectual in mutant B-RAF patients, combination of MEK inhibition and B-RAF inhibition is being explored. In patients with mutant B-RAF, dabrafenib and trametinib were combined and compared to single dabrafenib treatment. Not only was progression-free survival increased to 9.4 compared to 5.8 months, less secondary squamous cell carcinoma was observed (7–19 %) [38]. Another MEK inhibitor, cobimetinib, is also in clinical trial in combination with vemurafenib, in hopes to improve upon mutant B-RAF and MEK inhibition therapy (NCT01689519). In addition to MEK inhibition, ERK is also a potential target, as it lies downstream of the MAPK pathway. Small molecule inhibitors of ERK are in use, such as SCH772984, and another molecule BVD-523 is currently in clinical trial [89] (NCT01781429).
2.7 PI3K Pathway Members in Therapy
The phosphatidylinositol 3-kinase (PI3K) pathway is activated by multiple growth factors and results in increased cell survival and proliferation (Fig. 2.2). After binding of growth factor to a receptor tyrosine kinase, PI3K is activated and in turn activates AKT, which regulates multiple downstream components to promote cell growth [75]. Multiple components of the PI3K pathway are found to be dysfunctional in melanoma. Loss of PTEN, a phosphatase and negative regulator of the PI3K pathway, has been found to be able to induce melanoma formation in concert with B-RAF mutation [23]. This pathway is also activated in cancer by mutations in AKT as well as amplifications of receptor tyrosine kinases such EGFR and c-KIT. There are multiple small molecule inhibitors currently being tested that target the PI3K pathway.
PI3K itself has several drugs in clinical trials for advanced metastatic disease, including SAR260301, XL147 (SAR245408), and pictilisib (GDC-0941). SAR260301 selectively inhibits the PI3Kβ isoform. Both XL147 and pictilisib directly bind isoforms of PI3K, effectively competing for ATP binding. However, the clinical efficacy for use of these drugs in melanoma has yet to be concluded.
AKT, a serine-threonine kinase downstream of PI3K, has been shown to be important for transformation to melanoma [45], and increased expression of phosphorylated AKT mirrors disease progression [22]. Increased AKT activity or by activating mutation (specifically AKT3) in melanoma can be due to either dysregulation of the PI3K pathway or by activating mutation and is observed in about half of nonfamilial melanomas [25, 104]. Thus, AKT is an attractive target for therapy. Initially, perifosine, an AKT inhibitor, was not found to exhibit a clinical response [33]. However, AKT targeting is still being explored, and MK2206 is currently in trial for advanced melanoma.
In addition to single-agent targeting of the PI3K pathway, it has been shown in melanoma cells that combination inhibition of the PI3K and MAPK pathways is a more efficient therapeutic [102]. GSK2141795, which is able to specifically bind and inhibit AKT, is being explored in combination with trametinib in uveal melanoma (NCT01979523). Additionally, a clinical trial combining the MEK inhibitor AZD6244 with the AKT inhibitor MK2206 is also proceeding (NCT01021748).
Mammalian target of rapamycin (mTOR) regulates cell proliferation and angiogenesis and is an upstream activator as well as a downstream target of AKT (Fig. 2.2). Temsirolimus is an inhibitor of mTOR and has been evaluated in phase 2 clinical trials in combination with other therapies. When temsirolimus is given to patients in combination with sorafenib compared to sorafenib with tipifarnib, no substantial difference was observed with either progression-free survival or overall survival [76]. While the downstream targets of the PI3K pathway have yet to be fully examined, upstream RTKs are also being studied.
2.8 Decreasing Melanoma Growth Through RTKs
c-KIT, encoded by the KIT gene, is found to be mutated in at least 2 % of melanomas [120]. Mutation or amplification of the KIT gene was found to be present in 28 % of melanomas with high sun exposure, as well as 36 % of acral and 39 % mucosal melanomas [21]. Stem cell factor (SCF) is the ligand for the RTK c-KIT and upon ligand binding c-KIT is able to activate both the MAPK and PI3K pathways. As c-KIT mutations are found in other cancer types, small molecule inhibitors have already been created and are being tested in melanoma.
Nilotinib (Tasigna, AMN107) is a small molecule c-KIT inhibitor that is FDA approved for use in chronic myeloid leukemia and currently undergoing trials for use in melanoma (Fig. 2.2). A small phase 2 clinical trial of nilotinib in patients with c-KIT alterations showed low toxicity and promising inhibition of tumor progression, with a decrease in tumor size in 44 % of patients. Interestingly, greater progression-free survival was observed in treated patients harboring c-KIT mutation (8.4 months) than those with amplifications (1.7 months) [18]. This trend was also observed with imatinib mesylate (Gleevec®, Glivec®) which showed a higher response rate for those harboring KIT mutations rather than amplification [49, 56]. A study with sunitinib, a general tyrosine kinase inhibitor, also showed a higher percentage of response in those with KIT mutations vs. amplifications [85]. All of these drugs are tyrosine kinase inhibitors with activity not only against c-KIT but also other tyrosine kinases as well, such as PDGFR, and more specific drugs may have different outcomes. Dasatinib, another tyrosine kinase inhibitor that also shows high selectivity for SRC in addition to c-KIT, was examined in patients with advanced melanomas. No substantial effect was seen in melanoma and was found to be relatively toxic [67] (Fig. 2.2).
In addition to c-KIT, the RTK ERBB4 is also being targeted in melanoma. The ERBB4 gene is found to be mutated in 19 % of patients with melanoma, which results in increased activity [93]. Lapatinib is a tyrosine kinase inhibitor that is already approved for use by the FDA in breast cancer. It is currently undergoing phase 2 trial in patients with advanced melanoma that are positive for ERBB4 mutations (NCT01264081).
2.9 Inhibition of Angiogenesis
In order for a tumor to grow over a few hundred micrometers in diameter, new blood vessels must be formed to supply the new tissue mass with oxygen and nutrients, a process termed angiogenesis [84]. Development of new vasculature from existing vessels is orchestrated through expression of multiple proteins including cell surface receptors as well as secretion of growth factors. All of these aspects are found deregulated in cancers, including melanoma [31]. Destroying the ability of a tumor to create new vessels to support itself is an attractive therapy that could halt tumor growth. Additionally, the leaky, unorganized vessels found in tumors also make drug delivery much less efficient. Antiangiogenic therapies have been proposed to cause tumor vasculature normalization, instead of complete collapse, which may enhance drug delivery and therefore would be more successful as a combination therapy [58].
Vascular endothelial growth factor (VEGF) binding to its receptor VEGFR results in enhanced angiogenesis through endothelial cell proliferation as well as increased vessel permeability. VEGF and its receptor are overexpressed in melanoma, and inhibitors of this ligation are being explored in therapy. Bevacizumab is a humanized antibody that binds VEGF-A and inhibits receptor association and is currently FDA approved for use in colorectal cancer. Trials with bevacizumab in advanced melanoma include combination with other therapeutics. In a phase 2 trial, bevacizumab was combined with the mTOR inhibitor everolimus, and an average 4-month progression-free survival time and 8.6-month overall survival time were observed [51]. These results show that MAPK pathway inhibition combined with bevacizumab could be a promising therapeutic. Combination of bevacizumab with temozolomide resulted in an overall survival rate of 9.6 months, which was enhanced to 12 months if only considering B-RAF wt patients [114]. However, combination of bevacizumab with fotemustine showed an overall survival time of 20.5 months [26]. A large clinical trial including 214 patients compared treatment of paclitaxel and carboplatin with or without bevacizumab addition. Bevacizumab increased overall survival time to 12.3 months as compared to 9.2 months, and while the trend appeared to favor bevacizumab addition, it was not statistically significant [64]. While angiogenesis is an important part of tumor growth, the lack of compelling responses may be due to the fact that all these trials were given to late stage patients, at a point when new vessel formation is not as critical.
2.10 Immunotherapy
Normally the immune system works to clear pathogens and damaged cells; however, cancer cells have found a way to evade destruction and thrive. Many therapies being developed against cancers have been aimed to initiate clearance by the immune system. This is particularly true in melanoma, which is considered a highly immunologic tumor. High levels of tumor-infiltrating lymphocytes observed within tumor sites, as well as documented cases of spontaneous regression, have contributed to this label [19, 62]. Many mechanisms by which tumor cells evade immune system recognition are being targeted in melanoma and will be discussed below.
For T-cell activation during the immune response, a receptor presented on the surface of T cells, CD28, will bind a co-stimulatory molecule, B7, on the antigen-presenting cell (APC), in addition to a T-cell receptor binding antigen presented by the major histocompatibility complex (MHC) (Fig. 2.4). However, if cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is also presented on the T cell, it will compete with CD28 for B7 binding and inhibit T-cell activation, resulting in inhibition of cytokine production and T-cell proliferation [13]. Therefore, inhibition of CTLA-4 promotes T-cell response and enhances immune system response. Ipilimumab is a fully human IgG1 monoclonal antibody that targets and blocks CTLA-4 and is FDA approved for use in melanoma. In clinical trials, ipilimumab administered with gp100 peptide vaccine vs. vaccine alone produced an overall survival of 10 months compared to 6 months, respectively [57]. Furthermore, these results were supported by another clinical trial that compared ipilimumab in combination with dacarbazine to dacarbazine alone. Overall survival rates were increased to 11.2 months when ipilimumab was added, from 9.1 months of dacarbazine alone. Additionally, 20.8 % of patients treated with the combination were alive after 3 years, compared to 12.2 % of chemotherapy alone [97]. While response rates are low, the lasting results achieved by ipilimumab show promise for future immunotherapy, and it would be important to characterize the propensity for response.
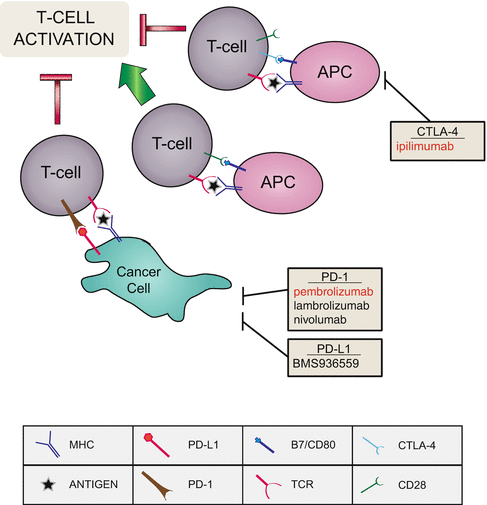
Fig. 2.4
Immune signaling being exploited in melanoma therapy. In hopes to increase immune response to melanoma, receptors and ligands that are deregulated in melanoma are being targeted to increase T-cell activation. Red writing indicates FDA approval in melanoma. APC antigen-presenting cell, MHC major histocompatibility complex, TCR T-cell receptor
In addition to CTLA-4, another inhibitory receptor expressed on activated T cells is programmed death-1 (PD-1), although it is mostly active during chronic inflammation (Fig. 2.4). Upon binding of PD-1 to its ligands (PD-L1 or PD-L2), T-cell response is reduced and apoptosis of the T cell can be induced. Notably, many cancers, including melanoma, have been found to overexpress PD-L1, which lends to tumor evasion [27]. Given the promise of ipilimumab, antibodies blocking PD-1 and PD-L1 are also being tested in melanoma. Blocking PD-L1 accessibility by treatment with a PD-L1 targeting antibody (BMS-936559) was explored in a 52 patient cohort. Of these patients, nine exhibited an objective response, and three achieved complete response [12]. Lambrolizumab (MK-3475) is an anti-PD-1 antibody that has shown effectiveness in patients with advanced melanoma, with a response rate of 25–52 %, depending on dosage [52]. Nivolumab (MDX-1106) is a genetically engineered fully human IgG4 monoclonal antibody specific for human PD-1. Phase 1 clinical trials have used nivolumab in combination with ipilimumab to block both CTLA-4 and PD-1 in advanced melanoma. High occurrence (at least 88 % of patients) of adverse events, including rash, fatigue, and diarrhea, was observed. However, these were generally reversible with treatment of immunosuppressants. In their treatment group that consisted of both nivolumab and ipilimumab for 3 weeks followed by nivolumab alone for 3 weeks, a clinical response was observed in 65 % of patients. Not only was their response rate higher than that observed previously for either treatment alone, but 31 % of patients had at least an 80 % reduction in tumor size at 12 weeks [121]. Overall, targeting CTLA-4 and PD-1 shows not only a high response rate but also a relatively high durable response.
The most recently FDA approved drug for melanoma is a PD-1 antagonist, pembrolizumab. In a phase 1 trial, patients with advanced melanoma who had already undergone treatment with ipilimumab achieved an overall response rate of 26 % at 8 months [126, 127]. These promising results, along with low toxicity, resulted in approval of the first PD-1 inhibitor by the FDA, to be used in patients with advanced disease that are not responding to other therapies. This approval emphasizes the promise immunotherapies hold, and identifying molecular markers of responding patients will provide more effectual results.
Adaptive cell therapy is another strategy being employed due to the immunologic nature of melanoma. In this approach antitumor-infiltrating lymphocytes are isolated and expanded ex vivo, then infused back into lymphocyte-depleted patient along with interleukin-2. Lymphocyte depletion by chemotherapy before injection of the expanded antitumor lymphocytes increased response rates from 49 to 72 %, with about half of patients still alive at 30 months [98]. Future trials aim to select tumor-infiltrating lymphocytes that are more effective. Results have shown that patients who achieve a complete response from adoptive cell transfer have a higher proportion of T cells that are CD8 positive and that also express the co-stimulatory molecule BTLA (B and T lymphocyte attenuator) [94].
2.11 Targeting ER Stress and Apoptosis
While multiple oncogenic pathways are disrupted in melanoma that result in increased proliferation, such as the PI3K and MAPK pathways, melanoma cells have also found a way to enhance their survival by resisting apoptosis. As already mentioned, the loss of CDKN2A locus expression results in two means the cell is able to overcome cell checkpoint control, thus thwarting apoptosis induction. However, that is not the only means melanomas escape death.
After protein translation, amino acid chains are brought into the endoplasmic reticulum (ER) for proper folding. However, under certain cellular insults that disrupt ER homeostasis, too much unfolded protein can accumulate in the ER, a condition known as ER stress. These protein aggregates can be very toxic to the cell, and ER stress can result in apoptosis [108]. ER stress can be compensated for by activation of the unfolded protein response, which can either halt protein synthesis or increase chaperone protein expression to help fold nascent proteins. During cancer progression and tumor growth, cancer cells experience hypoxic and nutrient deprived conditions, and this can trigger ER stress. In order to circumvent ER stress-induced apoptosis and survive, cancerous cells have found ways to either augment protein folding or sabotage apoptosis mechanisms. Impairment of a cancer cells ability to prevent apoptosis is a compelling therapeutic avenue, which could result in completion of apoptosis as well as render cancer cells more sensitive to cytotoxic drugs.
During the unfolded protein response, expression of chaperone proteins that help correctly fold proteins, such as heat shock protein 90(HSP90), is increased. It is also found that HSP90 expression is increased along with melanoma progression [8]. Additionally, oncogenic proteins are highly dependent on HSP90 for correct folding; this includes mutant B-RAF, making it an even more attractive therapeutic target [47]. Two inhibitors of HSP90 are currently undergoing clinical trials in advanced melanoma, XL888 and ganetespib (NCT01657591, NCT01551693) (Fig. 2.5).
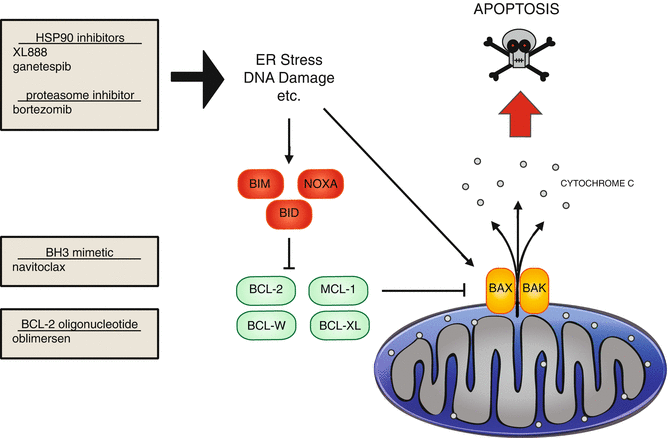
Fig. 2.5
Apoptosis signaling and experimental therapeutics. In response to multiple stimuli, including ER stress and DNA damage, apoptosis can be induced. Apoptosis is regulated by multiple BH3 domain containing proteins, which are divided into three classes. The green, antiapoptotic proteins, contains four BH3 domains. The other classes are both proapoptotic. The BH3 only proteins are indicated in red, and the proteins containing three BH3 domains are in orange. HSP90 inhibitors and proteasome inhibitors can induce ER stress and apoptosis. The BH3 mimetic navitoclax inhibits BCL-2, BCL-W, and BCL-XL. Oblimersen targets BCL-2
Another way to induce apoptosis through ER stress has been explored through the use of bortezomib, a proteasome inhibitor [55] (Fig. 2.5). Inhibition of the proteasome causes accumulation of proteins, including NOXA, and results in apoptosis. Bortezomib is currently FDA approved for treatment of mantle cell lymphoma and multiple myeloma and is being explored for efficacy in melanoma. However, bortezomib alone did not have a significant clinical effect in melanoma [77].
The intrinsic apoptosis pathway is triggered through formation of pores on the mitochondrial membrane, resulting in release of cytochrome c into the cytosol, generating a cascade of proteolysis (Fig. 2.5). These pores are either created or disrupted by BCL-2 protein family members, which are able to oligomerize with each other. There are three classes of proteins within the BCL-2 family, depending on their function (pro- or antiapoptotic) and basic homology (BH) domains [87].
1.
One class of proteins contains four BH domains (BH1-4) and is anti- apoptotic; examples include BCL-2, BCL-XL, BCL-w, BFL-1/A1, and MCL-1 (Fig. 2.5, green).
2.
The second class of proteins expresses three BH domains (BH1-3), is pro- apoptotic, and includes BAX and BAK (Fig. 2.5, orange).
3.
 Concept and Scientific Background of Personalized Medicine
Concept and Scientific Background of Personalized Medicine
 Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
 Personalized Management of Atopic Dermatitis: Beyond Emollients and Topical Steroids
Personalized Management of Atopic Dermatitis: Beyond Emollients and Topical Steroids
 Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
 Personalized Treatment in Cutaneous T-Cell Lymphoma (CTCL)
Personalized Treatment in Cutaneous T-Cell Lymphoma (CTCL)
 The Personalized Treatment for Urticaria
The Personalized Treatment for Urticaria




Related posts:
Concept and Scientific Background of Personalized Medicine
Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
Personalized Management of Atopic Dermatitis: Beyond Emollients and Topical Steroids
Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
Personalized Treatment in Cutaneous T-Cell Lymphoma (CTCL)
The Personalized Treatment for Urticaria
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
