Rachel S. Klein, Elisha Singer, Jacqueline M. Junkins-Hopkins, Carmela C. Vittorio, Alain H. Rook and Ellen J. Kim Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. J Am Acad Dermatol 2003; 49: 1049–58. The higher association with malignancy may represent selection bias.
Lymphomatoid papulosis
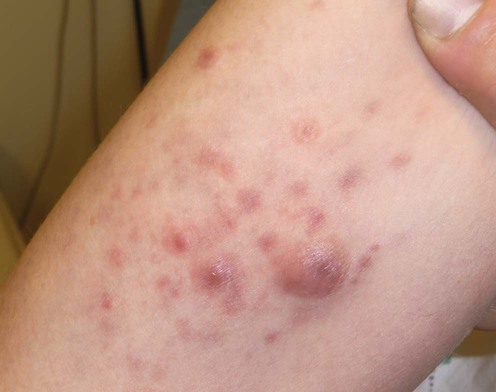
Specific investigations
CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma.
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree





