Mouse
Human
Unique
Shared
Unique
Shared
ILC1
NK
IL-12Rβ2
CD127 (not on NK)
CD90 (not on NK, ILC1)
CD117 (not on NK, ILC1, NCR- ILC3)
IL-12Rβ2
CD127 (not on some NK)
IL-1R (all but NK)
NKp46, NKp44 (on NK and NCR+ ILC3 only)
ILC1
IL-12Rβ2
IL-12Rβ2
ILC2
ST2, IL-17RB, ICOS, CD25
CRTH2, ST2, IL-17RB
ILC3
LTi
CD4, IL-23R
IL-23R, CD117
NCR+ILC3
IL-23R
NCR-ILC3
ND
ND
Group 1 ILC
ILC1s produce the Th1 signature cytokine IFN-γ and express the transcription factor T-bet. They include conventional NK cells as well as several recently-discovered IFN-γ-producing ILC1 subsets in the tonsils and intestines. ILC1 have also been found in human skin.
Conventional NK cells are the prototypical members of Group 1 ILC as they produce large amounts of IFN-γ. They co-express the T-box transcription factors T-bet and eomesodermin (Eomes), which, together with E4bp4 (Nfil), is required for their development and/or maturation [86]. All NK cells depend on IL-15 signaling for their development, survival and maintenance. Mouse NK cells are identified by their expression of the natural cytotoxicity receptors (NCRs) NKp46, and in certain mouse strains, NK1.1. Mature CD11bhiCD27lo and immature CD27hiCD11blo NK subsets are found in the spleen and bone marrow, whereas “tissue-resident” NK subsets are found in the liver, salivary glands, skin and uterus. In addition, there is a population of thymic-derived CD127+ NK subset in mice. In humans, NK cells are identified by CD56 and CD16 expression. Both CD56brightCD16− and CD56dimCD16+ NK cells are found in the blood and tissues. The CD56dim NK cells are highly cytotoxic and release perforin and granzyme upon encountering target cells, whereas CD56bright NK cells specialise in IFN-γ production in response to IL-12 and IL-18 [13]. NK cells also express germ line-encoded inhibitory and activating receptors of the C-type lectin-like family and immunoglobulin superfamily, which include the killer inhibitor receptors (KIR) (e.g., the Ly49 and CD158 series of molecules) and activating receptors (e.g., NKG2D, NKG2c) that control NK cell licensing and proliferation [46].
An additional ILC1 population, distinct from NK cells, was identified in 2013, in the tonsils and gut mucosa [6, 22, 45]. These ILC1 also express T-bet and NK cell-associated markers and produce IFN-γ but are developmentally and/or functionally distinct from NK cells. In particular, ILC1 derive from an Id2-expressing progenitor that does not give rise to NK cells and are dependent upon GATA-3 for their development, in contrast to NK cells. Phenotypically, non-NK ILC1 cells lack surface expression of the NK cell-specific Ly49 receptors, and possibly death receptors. Most ILC1s, including NK cells, also express the surface receptor IL-12Rβ2, consistent with their responsiveness to IL-12 stimulation.
One of the human non-NK ILC1 populations identified by Fuchs et al. within human tonsils was the NKp44+CD103+ subset [22]. The NKp44+CD103+ cells also secreted high amounts of IFN-γ when stimulated by IL-12 and IL-15, producing similar levels to that produced by blood CD56hi conventional NK cells. Some of these NKp44+CD103+ cells expressed perforin and granzymes and could degranulate, but unlike NK cells, they did not respond to IL-18 stimulation. These cells were also distinct from ILC3 (see below) as they did not respond to IL-23 stimulation, and they expressed higher levels of T-bet and lower levels of RORγt and the aryl hydrocarbon receptor (AhR) when compared to ILC3. Importantly, these cells were conserved in Rorc −/− and Ahr −/− mice which lacked ILC3. The murine counterparts of the NKp44+CD103+ subset were identified as CD160+NKp46+NK1.1+ cells in the intestine and tonsil. These cells were absent in Nfil3- or T-bet-deficient mice but still present in IL-15Rα-deficient mice, in contrast to conventional NK cells. Non-NK ILC1 localized specifically in the epithelial layer of the tonsils and in the intraepithelial layer of the small intestine, and had a phenotype that resembled tissue-resident memory CD8+ T cells (i.e., high levels of αEβ7 integrin and CD49a).
Independently, Bernink et al. [6] identified a population of cells in human tonsils that expressed T-bet and produced IFN-γ when stimulated with IL-12. After human ILC2 (CRTH2+) and ILC3 (cKit+NKp44+) were excluded from their analyses (discussed below), two additional populations were observed in the tonsils: the cKit+NKp44− and cKit−NKp44−. The cKit−NKp44− population produced IFN-γ but was distinct from the ILC1 population identified by Fuchs et al.: these cells did not express NKp44 and localized to the lamina propria instead of the intraepithelial layer of the small intestine. These cells were also distinct from NK cells, as they did not express perforin and granzyme, nor did they express the NK cell markers CD16, CD94 and IL-15Rα. Interestingly, Bernink et al. observed that a portion of these ILC1 could differentiate from cKit+NKp44−RORγt+ ILC3 precursors that had been isolated from both foetal gut and tonsil when they were stimulated with IL-2 and IL-12. This plasticity of ILC1 development from ILC3 to form an “ex-RORγt” subset was consistent with that reported in a RORγt fate-mapping study [95], where ILC3 cells in mice were shown to convert into IFN-γ producers (potentially ILC1). Therefore, there is some degree of plasticity between the ILC1 and ILC3 subsets, and the level of T-bet expression was suggested to promote the ILC3 to ILC1 differentiation (see later). Nonetheless, there also remains a separate ILC1 population that develops independently of RORγt. Notably, no ILC1 were found in the foetal gut, suggesting they may develop after colonisation with commensals.
Another group of researchers identified non-NK ILC1 after investigating the role of T-bet in ILC3 development [45]. They found that a third of NCR− ILC3 and almost all NCR+ ILC3 in the lamina propria expressed T-bet (see later). They also described a population of CCR6−RORγt+T-bet+ ILC that differentiated from the CCR6−RORγt+ ILC. These cells emerged postnatally under control of the aryl hydrocarbon receptor (AhR) in response to environmental stimuli (presumably commensal microbiota) and IL-23 stimulation. The increased expression of T-bet in this population in turn promoted expression of IFN-γ and NKp46.
Group 2 ILC
ILC2 produce the Th2 cytokines IL-5, IL-13, as well as IL-4, IL-9 and amphiregulin in response to IL-25 and IL-33, which may be further augmented by TSLP. ILC2 depend on the transcription factors GATA-binding protein 3 (GATA3) and the RORα for their development and function [35, 58, 99]. What is now known as ILC2 were first observed in 2001 as a non-B/non-T population that produces IL-5 and IL-13 in response to intranasal IL-25 administration [21]. In 2006, a population of Lin−cKit+CD90.2+ cells was described in the context of helminth infection in mice, whereby IL-25-dependent expulsion of Nippostrongylus brasiliensis occurred independently of B and T cells [20]. However, it was not until 2010 that ILC2 cells were properly phenotyped and functionally characterised, in which they were identified as “natural helper” cells [60], “nuocytes” [61] or “innate helper” cells [69], before the consensus term ILC2 was coined.
Murine ILC2 consistently lack expression of lineage markers CD3, B220, CD11b, Ter119 and Gr-1 [60, 69]. They express CD127 and the IL-17BR (IL-25R) and ST2 (IL-33R) receptors, consistent with their ability to respond to IL-25 and/or IL-33, respectively. Murine ILC2 cells, defined as a Lin−Sca−1+Thy1.1+CD127+T1/ST2+ population, were first found in the mesenteric fat-associated lymphoid clusters, mesenteric lymph nodes, spleen, liver and intestines of mice, and have since been detected in the bone marrow, liver, blood, airways and the skin. There is some variability in expression of other markers on ILC2, which may be due to the activation status and tissue-specificity of ILC2. For instance, lung ILC2 express lower expression of Sca-1 and CCR9 compared to those from the mesenteric lymph nodes (mLN); and skin ILC2 express CD103. Murine ILC2 also express the high-affinity IL-2 receptor alpha chain (IL-2Rα, CD25) [73], ICOS, and variable levels of major histocompatibility class II molecules and c-Kit (CD117) [33, 53, 54, 61], consistent with the fact that they do not rely upon stem cell factor (SCF) signaling for survival [60, 69, 72]. ILC2, in contrast to the other ILC subsets, do not express CD2, which further serves to discriminate ILC2 cells from conventional NK cells and most T cells [72]. ILC2 also lack the NK and ILC1-associated markers NK1.1 and NKp46, and unlike ILC1 and ILC3, they have not been shown to develop into other ILC.
The human ILC2 equivalents were first identified in the foetal gut and lung tissue [58] as a Lin−CD127+CD45hi population that also expressed transcripts encoding IL-13, IL-17RB, ST2 and CRTH2 but low levels of RORγt. ILC2 have also been identified in human blood, skin, lungs and nasal mucosa. In humans, ILC2 cells are identified by lack of expression of lineage markers CD3, CD19, CD94, CD1a, CD11c, CD123, BDCA2, CD14, FceR1 and CD34 and their expression of CD127, the pan-human-ILC marker CD161 and, uniquely, CRTH2 [58]. Similar to murine ILC2, human ILC2 also express CD25 [59] and variable levels of c-Kit, but do not express NKp44. Human ILC2 cells, like their murine counterparts, are also responsive to IL-25, IL-33 and TSLP, indicative of functional receptors to these cytokines [55, 57, 58, 89].
Group 3 ILC
ILC3 depend on the transcription factor RORγt and produce IL-17 and/or IL-22 in response to IL-1β and IL-23 stimulation [34, 82]. ILC3 are a complex family. The prototypical member is the lymphoid tissue-inducer (LTi) cell that is crucial for lymphoid organ formation during embryogenesis (Peyer’s patches, lamina propria) and in the neonate (cryptopatches and isolated lymphoid follicles), and that also has a role in lymphoid organ repair after infection [51, 52]. These cells express CD127, CD117, α4β7, CD4 and CCR6 in mice, although a population of CD4− LTi also exists. Two other non-LTi ILC3 subsets, distinguished as those that express natural cytotoxicity receptors (NCR) and those that do not, were also identified in nonlymphoid tissues, i.e., the gut mucosa and tonsils [10, 14, 50, 75, 77] where they are important for barrier protection. They include CCR6+CD4− cells and the CCR6-/lo population that are rare in the foetal liver but expands rapidly during the first month of life dependent upon the expression of AhR. The CCR6−/lo ILC3s are further subdivided into an NKp46− and NKp46+ subsets, i.e., NCR− ILC3 and NCR+ ILC3.
The NCR− ILC3 cells do not express IL-17 or CD4 but produce IL-22. The subset now collectively termed NCR+ ILC3 were also known as NCR22 cells, NKp46+ ILC, ILC22s and NKR-LTi. Although these cells expressed NKp46, they are functionally distinct from NK cells as they do not express cytotoxic molecules such as perforin, granzymes and death receptors. These cells also did not produce IFNγ or TNFα, but instead expressed IL-22. They are also developmentally distinct from NK cells, as fate-mapping experiments using RORγt-reporter mice showed that many of these cells had a history of RORγt expression whilst NK cells never did [50, 76, 77].
ILC Development, Diversification and Plasticity
The lineage-specific development, function and maintenance of ILC depend on a restricted set of transcription factors. Strikingly, these are the same set of transcription factors, i.e., T-bet, GATA-3 and RORγt, that are expressed by distinct CD4+ T helper (Th) cells, to coordinate the developmental and functional diversification of Th1, Th2 and Th17 subsets (Fig. 3.1). These transcription factors regulate a similar gene expression program in ILC as they do in T cells, resulting in unique developmental programs, which endow the individual ILC subsets with their specific effector functions. This striking parallel has led to the notion that ILC represent innate counterparts of Th cells, and are important for early immune defence, whilst setting the stage for coordination of the cytokine milieu between innate and adaptive immunity.
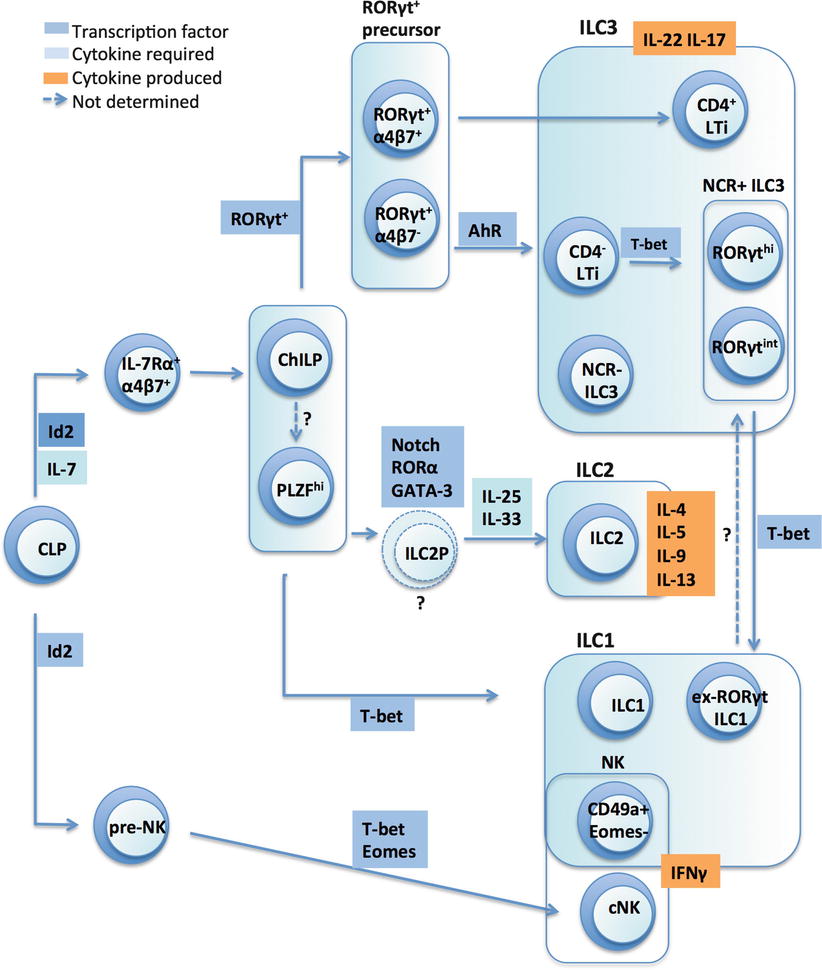
Fig. 3.1
ILC development is orchestrated by transcription factors. Inhibitor of DNA binding 2 (Id2), Retinoid-related orphan receptor (ROR), eomesdersmin (Eomes), Aryl hydrocarbon receptor (AhR), conventional NK cell (cNK), common helper-like innate lymphoid cell precursor (ChiLP), ILC2 precursor (ILC2P)
ILC are derived from the common lymphoid progenitor (CLP), which is derived from a pluripotent haematopoietic stem cell that has successively shed erythroid and myeloid potential. All ILC also rely on the common γ-chain (γc) used by various cytokine receptors and show a developmental requirement for the transcriptional regulator Id2 (inhibitor of DNA binding 2). Accordingly, genetic ablation of Id2 cripples the development of all known ILC lineages [100]. ILC are also absent in γc-deficient mice [76]. Expression of Id2 in the CLP is thought to inhibit its developmental potential of CLP into T and B cells while favouring ILC generation [35].
Earlier work had identified CLPs that expressed the integrin α4β7 in the foetal liver that were able to differentiate into NK cells and all subsets of RORγt+ ILC [68]. The severe reduction of α4β7+ CLPs in Id2-deficient mice indicates that Id2 lies upstream of Rorγt in the developmental pathway [11]. Hoyler et al. had also previously identified a precursor of ILC2 cells, termed ILC2P. Clues to the identity of the ‘elusive’ common ILC precursor was recently further revealed. The use of the Id2 reporter mouse by Klose et al. led to identification of a rare Id2+α4β7+Lin−IL-7Rα+ population in the bone marrow, which they termed ChILP (for common helper ILC precursor) [44]. Whilst the ChILP expresses the integrin α4β7, it does not express Flt3 or CD122, the recently described NK progenitor marker. It also does not express CD25 and may hence be distinct from the CD25+IL-33R+ ILC2P described by Hoyler et al. [35]. In adoptive transfer studies, the ChILP gave rise to ILC2 cells and ILC3 cells (including both LTi and NKp46+ ILC3 subsets), as well as an unusual non-NK ILC1 subset (which was CD49a+NKp46+T-bet+IL-7Rα+ but did not express Eomes). Notably, the ChILP did not give rise to conventional NK cell subsets.
Bendelac and colleagues created PLZF (promyelocytic zinc finger) fate-mapping reporter mice, chiefly to pursue their interest in NKT development [12]. As predicted, they found that NKT cells expressed GFP and were also ‘fate-mapped’, but, unexpectedly, also discovered a large fraction of GFP− ILC that were fate-mapped, i.e., had expressed PLZF at some stage during their development. They showed that there was a PLZFhi population within the α4β7+Lin−IL-7Rα+ progenitor cells that gave rise to ILC2 cells, mucosal CD4− ILC3 cells, and the same peculiar NKp46+CD49a+ ILC1 liver subset described by Diefenbach and colleagues [44]. Therefore, both the ChILP and the PLZFhi progenitors were α4β7+Lin−IL-7Rα+ and generated ILC2, ILC3, and the peculiar hepatic NK subset (now thought to correspond to tissue-resident NK cells), but did not give rise to conventional NK cells, suggesting they may be overlapping populations. Indeed, a large fraction of ChILP express PLZF. On the other hand, PLZFhi precursors also express Id2 and could be differentiated from PLZF−α4β7+Lin−IL-7Rα+ cells in vitro. Since the ChILP generates a broader range of ILC3 cells (including CD4+ LTi cells), it may be possible that the PLZFhi ILC precursor is a subset of the ChILP population but this remains to be determined.
Downstream of this committed ILC progenitor, the induction of GATA3, RORα, T-bet and RORγt transcription factors, together with contributions from the Notch signalling pathway and additional transcription factors such as TCF-1 and TCF-7, governs the polarization of individual ILC subsets (described below and recently reviewed by Tanriver and Diefenbach [87]).
NK cells and ILC1
Unlike ILC, NK cells do not require RORγt or IL-7 for their development but are dependent upon IL-15. There are several developmentally distinct NK subsets, including bone marrow and thymic-derived NK cells, conventional and tissue-resident subsets, that are developmentally driven by the transcription factors Tbx21, and Eomes, and as recently determined, E4bp4 (Nfil3), which promotes transcription of Eomes and Id2 GATA-3 [36]. A pre-pro NK cell (Lin−CD122+NK1.1−CD49b−Id2−GFP+) was also recently identified in the Id2-GFP reporter mouse [9]. Eomes is up-regulated during the transition of CD49b− immature NK cells to CD49b+ mature NK cells during which they acquire effector function [26]. In contrast, T-bet expression is already found in immature NK cells and T-bet regulates the egress of mature NK cells from the bone marrow [91]. Recently, two groups identified a subset of liver CD49a+ NK cells that developed independently of Eomes and E4bp4 (Nfil3), and that were thus distinct from ‘conventional’ Eomes+ NK cells [15, 80]. This peculiar liver NK population was also generated from committed ILC precursors identified by Bendelac’s and Diefenbach’s groups and further studies might reveal if there are additional NK cell-restricted ILC precursors [12, 44].
From their investigations of ILC3 development using RORγt fate-mapping, Klose et al. identified CD127+ ILC1s, primarily found in the intestine, that are distinguished from bona fide NK cells [45]. Hence, whilst all IL-15Rα+ NK cells are RORγt fate map negative (RORγtfm-) and did not have a history of RORγt expression, two additional CD127+NK1.1+NKp46+ ILC1 subsets were identified as either RORγtfm+ or RORγtfm-. In humans, Fuchs et al. further identified a subset of intraepithelial ILC1s (CD3−CD56+NKp44+CD103+) that expressed T-bet but did not express RORγt. Their development required expression of E4bp4 and T-bet. This subset produced IFN-γ after stimulation with IL-15 and IL-12, displayed some cytotoxic activity, but was still present in IL-15Rα-deficient mice. Bernink et al. identified another ILC1 subset (CD127+Kit−NKp44−) that resided in the lamina propria of the intestine and expressed T-bet and low levels of RORγt mRNA [6]. Interestingly, cells with this phenotype could also be derived from the RORγt+ ILC3 upon IL-12 stimulation. These “ex-RORγt+” cells indicate that there exists some developmental plasticity between ILC3 and ILC1, which has not been described for ILC2.
ILC2
are developmentally dependent upon on the transcription factors Id2 [35, 60], GATA-3 [35] and RORα [28, 29, 97]. In contrast to ILC3s, ILC2s are not well represented in the newborn gut but are generated in the bone marrow. An ILC2 precursor (ILC2P) was identified in earlier studies in the bone marrow, which is defined as Lin−Sca-1hiId2hiGATA-3hi (‘LSIG cell’). This precursor expressed IL-33R and IL-17RB but lacked KLRG1 expression. IL-33 stimulation could convert these cells into strong producers of IL-5 and IL-13. GATA-3 is required for the differentiation of ILC2Ps, as well as the peripheral maintenance of mature ILC2s [35]. Furthermore, two recent studies revealed the additional functions of GATA-3 in dose-dependent control of ILC2 development and intracellular phosphorylation pathways [23, 43]. ILC2 also express high levels of RORα, which is required for their development. Staggerer mice (Rora sg/sg), which carry a functional null mutation of RORα, have an intrinsic defect in the development of ILC2s and are also impaired in their ability to clear worm infection [97]. In addition to GATA-3 and RORα, T-cell factor 1 (TCF-1), acting downstream of Notch signalling, is also required for ILC2 development.
ILC3
depend on the transcription factor RORγt. ILC3 include LTi cells that are crucial for lymphoid organ formation [45, 95] and in the foetal liver, the Lin−α4β7+IL-7Rα+ progenitor differentiates into LTi precursors upon Id2 and Notch signaling [11]. The additional RORγt+ ILC3 population resides in the intestinal lamina propria and can be further subdivided into CCR6+CD4− cells and a CCR6-/lo population that expand in the intestines during the first month of life, requiring AhR expression. The CCR6−/lo ILC3s are further subdivided into NKp46− and NKp46+ subsets, i.e., NCR− ILC3 and NCR+ ILC3. In addition to RORγt, expression of T-bet is also important for the generation of some ILC3. Hence, T-bet is highly expressed by NCR+ ILC3 in the mouse lamina propria [45, 79], and together with TCF-1 is also involved in the differentiation of some NCR− ILC3 to NCR+ ILC3 [45, 70, 79]. It has been suggested that an increasing T-bet gradient controls the sequential upregulation of NKp46 and NK1.1 on NCR− cells with the concomitant downregulation of RORγt [45, 95]. Rankin et al. recently showed that T-bet was indeed an essential transcription factor essential for the NCR+ ILC3 population, as NCR+ ILC3 were absent in T-bet-deficient mice. The progenitor of LTi, NCR− ILC3 and NCR+ ILC3 had been a matter of controversy, but in the same study, Rankin et al. showed that NKp46+ ILC3 was derived only from the CD4− LTi population. Therefore, NCR+ ILC3 and CD4− LTi were distinct from CD4+ LTi and may be generated from bona fide Rorγt+α4β7− subset which also gives rise to a significant population of NKp46+ cells.
The lineage relationships between ILC populations and the developmental plasticity between ILC subsets that might be driven in response to environmental stimuli, is an exciting area of research. Recent information on the developmental pathways of mouse ILC has further led researchers to suggest that NK cells and CD127+ ILC could be considered innate forms of CD8 and CD4 T cells, respectively [31]. It is fascinating that the same conserved set of cell fate-determining transcription factors were ‘selected’ to drive the development and diversification of innate lymphoid cells, as well as T cells, their more contemporary counterparts.
ILC in the Healthy Skin
Three groups recently determined the composition of ILC subsets in human adult skin, which was found to harbour all the ILC subsets described to date [18, 89, 94]. Dyring-Andersen et al. analysed cells isolated from dermal explants that were cultured in IL-2 for 11 days [18]. Since the lineage cocktail they used for exclusion of non-ILC did not include the NK cell markers, NK cells were also included in their analyses. The expression of CD56 and RORγt was used to delineate the ILC3, ILC2 and NK cell subsets. Hence, RORγt+CD56+ cells were identified as the NCR+ ILC3 population. This NCR+ ILC3 were also shown to express NKp44, NKp46 and CD117. The RORγt+CD56− cells that were CD117+ were termed NCR− ILC3, whereas RORγt−CD56− cells were identified as NK cells after the exclusion of ILC2 (which lacked NCR expression) within this population. In healthy skin, ILC2 and NCR− ILC3 were the most prevalent subsets and total ILC was estimated to constitute ~9 % of the CD45+ cells. In contrast, Teunissen et al. and Villanova et al. examined freshly prepared cells from whole skin, or cells prepared from separated dermis and epidermis, in addition to explant cultures. In both studies, skin ILC were first identified as CD45+Lin−CD127+ populations, thereby excluding most NK cells. The CD45+Lin−CD127hi ILC were reported to represent ~1.3 % of CD45+ cells. In both of these studies, skin ILC2 were identified by CRTH2 expression, and the remaining subsets were distinguished by CD117 and NKp44 expression: CD117+NKp44+ cells were identified as NCR+ ILC3 and CD117+NKp44− cells were identified as NCR− ILC3, whereas subsets that lacked expression of both CD117 and NKp44, but expressed CD161, were identified as non-NK ILC1. The ILC2, ILC1 and NCR− ILC3 populations were present in similar proportions in freshly isolated dermal cells. However, NCR+ ILC3 were notably rare in these samples. Instead, NCR+ ILC3 were present in significant proportions in dermal explant cultures, consistent with the representation of ILC3 in explants observed by Dyring-Andersen et al. Teunissen et al. further showed that isolated NCR− ILC3 could convert into NCR+ ILC3 when stimulated with IL-1β and IL-23 in vitro. This plasticity between ILC3 and ILC1 was similar to that reported for NCR− ILC3 isolated from human tonsils and foetal intestine, which could differentiate into NCR+ ILC3 after IL-1 and IL-23 stimulation [6]. Villanova et al. also showed that CD3−IL17+ cells could be found in both the dermis and epidermis of healthy skin, whereas CD3−IL-22+ cells could only be identified in the epidermis, but not the dermis [94]. Compared to the blood which contained <1 % of ILC, the skin was enriched for ILC. The blood also comprised ILC that had skin-homing potential, as ILC in healthy control blood expressed CLA at high frequencies (~33 %) compared to T cells (11–18 %). CLA expression was particularly high on the NCR− ILC3 subset. Thus, ILC could potentially migrate into and populate the skin, although the cues that guide this process are still unknown.
ILC2, ILC3 and NK cells have also been positively identified in the mouse dermis [40, 64, 72, 90]. ILC2 and ILC3 accumulation have been described in a few studies in the context of atopic dermatitis and psoriasis, respectively. As the skin ILC populations are only recently beginning to be mapped, some key functions of ILC in other tissues will be discussed in the following sections to provide insight into their potential roles within the skin.
Functional Specialisation of ILC and Their Potential Roles in the Skin
Group 1 Innate Lymphoid Cells
In addition to IFN-γ, NK cells produce cytotoxic molecules including granzyme A, perforin and death receptors, which are crucial to their key function of eliminating infected cells or tumours. Their function within the skin is less clear, and whether NK cells are involved in the pathogenesis of inflammatory diseases, such as psoriasis, remains controversial. NK cells have been detected in psoriatic skin [8, 63, 90], and are thought to exacerbate skin inflammation. Some leukocytes isolated from biopsies from psoriatic skin showed a CD56bright CD16− phenotype, which classifies them as either bona fide NK cells and/or ILC1 [63]. Nevertheless, a cell population that expressed the NK inhibitory receptors CD158b, CD94 and NKG2A, therefore more likely representing NK cells, were also found in psoriatic lesions [8]. In contrast, a recent study observed a substantially lower percentage of CD57+CD56+CD16+ NK cells in lesional psoriatic skin compared to control skin. NK cells were also decreased in the peripheral blood of psoriatic patients [49]. Some evidence in support of a role for NK cells in psoriasis pathogenesis came from a (SCID) mouse xenograft model, whereby NK cells from psoriatic, but not normal donors that were injected into autologous non-lesional human skin grafts could induce histopathology that resembled psoriasis. In summary, the role of NK cells in the skin during homeostasis and inflammation remains unclear.
The other ILC1 subsets are distinct from NK cells in development and function. These cells have been described in mucosal tissue, and the non-NK ILC1 subset has also been identified in human skin. Non-NK ILC1s express T-bet and produced IFN-γ upon IL-12, IL-15 or IL-18 stimulation [6, 22, 45]. These ILC1 populations include the NKp44+CD103+ cells first described within human tonsils. Their murine counterparts reside in the intraepithelial layer of the tonsil and small intestine. Based on their tissue localisation and their phenotype, which resembles tissue-resident memory CD8+ T cells (TRM; αEβ7hiCD49ahi) that are observed after viral infection, the ILC1 cells identified in human skin might be viewed as the innate counterparts of TRM which are poised for prompt effector functions. It is possible that epidermis-associated ILC1 might provide early responses to stress or infection. Notably, some of these ILC1 bear hallmarks of TGF-beta imprinting (i.e., they express CD103, CD9 and NEDD), suggesting that skin ILC1 might be modulated by this cytokine, similarly to Langerhans cells.
The second subset of ILC1 was identified by Bernink et al. in human tonsils, and in the intestinal lamina propria, but was not localized to the epithelium [6]. These cells also produced IFN-γ in response to IL-12 (instead of IL-18 and IL-15) but were not cytotoxic. The third description of ILC1 was by Klose et al., who identified a population of CCR6−RORγt+T-bet+ ILC1 that differentiated from the CCR6−RORγt+ ILC. In the gut at least, these ILC1 may impact protection and pathology. For instance, CCR6-RORγt+ ILC that produced IFN-γ mediated protective immunity against Salmonella typhimurium infection by promoting mucus secretion. These early IFN-γ responses might sensitize antigen-presenting cells and shape the local microenvironment to influence subsequent effector T cell responses. Conversely, ILC1 can also contribute to intestinal pathology. Both the intraepithelial CD56+NKp44+CD103+ subset and the CD56−c-Kit−NKp44− subset in the lamina propria were increased in the inflamed ileum mucosa of patients with Crohn’s disease. Murine ILC1 have also been shown to accumulate in two mouse models of gut pathology, i.e., DSS-induced gut inflammation in mice reconstituted with human PBMC, and in an innate immune-mediated colitis model, in which RAG−/− mice were administered agonistic anti-CD40. Whether they have similar dual roles in the skin remains to be elucidated.
Group 2 ILC Functions
ILC2 are found in the respiratory and gastrointestinal tissues as well as in skin. Whilst ILC2 were originally described as important in protective type 2 immunity against helminth infection, studies from mouse models of asthma and atopic dermatitis also suggest a role for ILC2 in promoting allergic inflammation.
ILC2 in Airway Allergic Inflammation
Allergic inflammation is characterized by increased expression of Th2 cytokines (IL-4, IL-5, and IL-13), resulting in tissue eosinophilia, mast cell degranulation, mucus production and IgE production. Although allergic responses are traditionally thought to be primarily driven by CD4+ Th2 cells, there is increasing evidence that ILC2 are also important, as they too produce significant amounts of IL-5 and IL-13, both constitutively and upon activation. ILC2 have been shown to contribute to type 2 lung inflammation in murine models of allergic airway inflammation induced by different allergens, including fungal [4], house dust mite, glycolipid [41], or ovalbumin [2, 28, 29, 42] in the absence of T cell-derived cytokines. In these models, ILC2 can be activated by epithelial-derived cytokines IL-25, IL-33, and TSLP to produce IL-5 and IL-13, as well as IL-9, effector cytokines which contribute to airway hyperreactivity, eosinophilia and mucus production. Under certain conditions, lung ILC2 are also able to produce IL-4, for instance, when stimulated with TSLP and leukotriene D4 [17]. Overall, these reports suggest that ILC2 can direct distinct pathogenic lung inflammatory responses depending on their various cytokines produced. On the other hand, ILC2 has been shown to produce the EGFR ligand amphiregulin to promote epithelial regeneration after allergen or viral challenge [17, 59]. Therefore, ILC2 can have dual roles in pathogenecity and repair in epithelial tissues, and the dysregulation or inappropriate activation of ILC2 are associated with allergic responses that are classically thought to be driven by CD4+ Th2 cells.
ILC2 in the Skin
Apart from the gastrointestinal tract, lymphoid tissue, lungs and airway mucosa, ILC2 have been identified in both murine and human skin. In murine skin, ILC2 preferentially localise in the dermis, where they constitute 5-10 % of all dermal CD45+ cells [72]. Murine dermal ILC2 are highly enriched in the skin compared to the blood and to other tissues (with the exception of fat tissue). Consistent with the developmental requirements of all ILC subsets, dermal ILC2 are absent in Id2−/−, IL7−/− and Rag-1−/− mice. Interestingly, dermal ILC2 were enriched in RAG−/− mice, suggesting that they may share and compete for survival factors with T cells that are otherwise present in wildtype murine skin.
The ILC2 in murine dermis differ slightly from those found in other organs: they express CD103, which recognizes E cadherin, and they are c-Kit negative, suggesting that there are local factors that dictate ILC2 phenotype and function in the skin. Dermal ILC2 also express CXCR6, which enabled Roediger et al. to capture ILC2 behaviour in the dermis of CXCR6-eGFP+/− mice using intravital multiphoton microscopy [72]. The dermal ILC2 cells displayed a patrolling movement which was slower compared to T cells. They were also observed to make prolonged contacts (of up to 30 min) with mast cells. Functionally, during steady-state, ILC2 were found to be the primary contributors to homeostatic IL-13 production in the skin. Roediger et al. further found that IL-9 enhanced mast cell release of IL-6 and TNF-α ex vivo, while IL-13 had the opposite effect and suppressed mast cell release of IL-6 and TNF-α [72]. It was postulated that one of the functions of ILC2 in steady-state could be to suppress mast cell activation through constitutive IL-13 production.
ILC2 have also been identified in healthy human skin [40, 74, 89]. Human skin ILC2, like other ILC subsets, does not express the common leukocyte lineage markers. Human ILC2 express CD127 and CD161, and are further distinguished from other ILC by expression of CRTH2, ICOS, CD25, IL33R, TSLPR and IL-25R. Compared with the paucity of circulating ILC2 in the blood (<0.2 % of lymphoid cells), ILC2s are highly enriched in healthy human skin tissue (up to almost 3 % of lymphoid cells). Furthermore, a significant proportion of skin ILC2 expressed the skin-homing chemokine receptors CCR4, CCR10 and CLA, and thus may be distinct from circulating ILC2. Interestingly, Salimi showed that whilst human ILC2 do not express the NK cell-related KIRs, NKp46 and NKp44, they expressed KLRG1 [74]. The authors demonstrated that the interaction of E-cadherin, an adhesion protein important for maintaining epithelial integrity, with KLRG1 on ILC2 inhibited production of cytokines and amphiregulin in vitro. They proposed that expression of KLRG1 may provide a barrier-sensing mechanism, whereby the normal activity of ILC2 is dampened by keratinocytes or LCs that migrate through the dermis. Upon the loss or cleavage of E-cadherin during skin inflammation, such as that often observed during atopic dermatitis [92], ILC2 inhibition may be discontinued, resulting in their inappropriate activation [74].
The enrichment of ILC2 in the skin suggests they may have important roles in homeostasis and barrier defence. It has also been proposed that ILC2 may have a role in skin homeostasis, as they also produce amphiregulin, may participate in skin remodelling by stimulating keratinocyte proliferation and promoting wound healing responses. Studies that expand upon our understanding of their interactions with other skin-resident cell types would help us decipher their roles and how they contribute to immune responses and maintenance of barrier function in the skin.
ILC2 in Atopic Dermatitis
There have only been a few studies characterising ILC2 during skin inflammation, and these were performed in the context of atopic dermatitis (AD), a chronic inflammatory skin condition associated with skin barrier disruption, eosinophilic infiltration, and high serum IgE levels. This disease affects up to 30 % of children of the industrialized world and is precipitated by both genetic and environmental factors [19]. Barrier dysfunction is a key early event in the pathogenesis of AD which is further complicated by inflammation driven by type 2 cytokines. Indeed, null mutations in the filaggrin gene that is involved in barrier integrity are found in patients with severe AD [38].
The three key ILC2 activators, IL-25, IL-33 and TSLP, are produced by a variety of cell types found in the skin, including dermal fibroblasts, epithelial cells and myeloid cells. IL-25 is a member of the IL-17 family and has been associated with Th2-like inflammation and disease. IL-33 is a member of the IL-1 family that binds the ST2 receptors that are preferentially expressed on Th2 cells and a range of innate immune cells. IL-33 is involved in Th2 polarisation, and also acts as an alarmin when released during tissue necrosis, or when cleaved by caspase 1 during programmed cell death. TSLP is also thought to be involved in type 2 responses. It has been proposed that epithelial damage can lead to the production of these “initiating cytokines” (IL-25, IL-33, TSLP), which are potent activators of ILC2. Indeed, during AD, elevated levels of IL-25, IL-33 and TSLP, in addition to both pro-inflammatory and Th2 cytokines have been reported [78, 83].
ILC2 in humans also express CRTH2, the receptor for prostaglandin D2. CRTH2+ ILC2 isolated from human skin have been shown to respond to IL-33 and TSLP stimulation by secreting IL-13 [89]. ILC2 lines generated from the skin also produced IL-13 when stimulated by PMA/ionomycin, and TSLP could synergise with IL-25 to increase their production of IL-13. Interestingly, in one study, IL-33 did not further enhance IL-13 production by skin-derived ILC2 lines [40], which was somewhat different to the responses of human ILC2 isolated from nasal polyps and blood, in which IL-33 and TSLP synergistically enhanced type 2 cytokine production [55]. In contrast, Salimi demonstrated a hierarchy of human ILC2 responses upon ex vivo stimulation, with IL-33 being most potent at inducing cytokine production and ILC2 migratory behaviour [74]. Activation of CRTH2 by prostaglandin D2 has been shown to synergise with IL-25 and IL-33 to induce IL-13 release from circulating ILC2 [3]. Recently, Ogg and colleagues demonstrated that endogenous prostaglandin D2 released by mast cells could stimulate CRTH2+ ILC2 isolated from human skin to migrate and produce a variety of pro-inflammatory cytokines, in addition to IL-4, IL-5 and IL-13 [98]. Together, the results indicate that ILC2 primary cells or cell lines cultured from human skin can be regulated by TSLP, IL-25, IL-33 and prostaglandin D2. The differences between studies could indicate flexibility in ILC2 responses to different stimulators, or the presence of different subsets that may have intrinsically different cytokine profiles within the skin.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


