Fig. 1.1
LC and DC populations in human skin. (a) LC and DC residing in human skin in the steady state and (b) infiltrating the skin during inflammation. The main suface markers and pattern recognition receptors of the respective subpopulations are depicted
From a conceptual viewpoint, it makes perfect sense to assume that danger signals not reaching beyond the epidermis, i.e. the site of LC, will not mobilize all the armed forces the immune system is capable of activating. On the other hand, virulent microorganisms that breach the dermo-epidermal junction and thereby can reach DDC should trigger a massive host defense response that can successfully eliminate the pathogen. From this perspective, one would expect that LC mainly act as regulatory antigen-presenting cells (APC) inducing a state of antigen-specific non-responsiveness. Following this reasoning, DDC should remain inert under homoeostatic conditions and mature into potent sensitizing DC upon receiving appropriate activation signals. The “black and white” picture of the roles of LC vs. DC is probably not tenable under all circumstances. As an example, in response to an overwhelming microbial insult, LC can engage themselves in the promotion of a T effector cell response [7]. Although we have gained more insight into the LC and DC network in the skin, we are still far from understanding it in all its complexity. This is illustrated by the recent finding of another (although quantitatively minor) DC subset residing in steady-state skin, i.e. CD141+ DC (Fig. 1.1a) [11]. These DC seem to be mainly engaged in antigen cross-presentation [12].
In an inflammatory setting, skin-resident LC, DDC and CD141+ DC undergo phenotypic and functional changes. In addition, various other types of DC are entering the stage (cf. Fig. 1.1b). These blood-derived DC include the so-called plasmacytoid DC (pDC), various DC with an inflammatory phenotype (inflammatory DC, IDEC) and CD14+ (monocyte-derived) DC. All of them exhibit distinct features with regard to their antigen presentation properties and interaction with other immune cells (for review, cf. [13, 14]). They play major roles in the pathogenesis of various skin conditions such as atopic dermatitis (Chap. 22) and psoriasis (Chap. 21). Their involvement in these diseases will be discussed in the respective chapters of this book.
Toll-Like Receptors: Bridging Innate and Adaptive Immunity
Pattern Recognition Receptors: Sensing the Danger
Until quite recently, it was essentially unknown by which mechanisms danger signals (such as immunogenic haptens and microorganisms) trigger the activation and maturation of LC and other DC in the skin and ultimately initiate an adaptive immune response. A series of discoveries (for review, cf. [15, 16]) has shed new light on this issue by revealing that DC function and development are essentially modulated by innate immune receptors recognizing damage- or pathogen-associated molecular patterns (DAMP and PAMP; listed in Table 1.1) (see Chap. 2). Among this growing family of pattern recognition receptors (PRR), the so-called toll-like pathogen recognition receptors (TLR) have been particularly well investigated. Ten TLR have been described in humans so far (listed in Table 1.2). TLR can be broadly divided into two groups (extra- vs. intracellular). Extracellular TLR (TLR1, 2, 4, 5, 6) essentially recognize bacterial and fungal products. Briefly, TLR2 combined with TLR1 or TLR6 mostly recognizes motifs of gram-positive bacteria (e.g. lipoproteins, lipotechoic acid (LTA)), while TLR4 senses gram-negative bacteria-associated lipopolysaccharides (LPS). Bacterial flagellin is recognized by TLR5. The intracellular receptors TLR3 and TLR7-9 recognize mostly virus-derived nucleic acids, i.e. double-stranded RNA (dsRNA; TLR3), single-stranded RNA (ssRNA) (TLR7-8) and CpG oligodeoxynucleotides (TLR9).
Table 1.1
Pattern recognition receptors (PRR) and their principal ligandsA) Principal PRR families
Group of PRR | Examples of PRR | Principal PAMP/DAMP(s) |
|---|---|---|
Nucleotide-binding oligomerization domain (NOD)-like receptors (NLR)a | NOD1 (CARD4) NLRP1B (NALP1) NLRP3 (NALP3) | iE-DAP, GM-tripeptide Anthrax letal toxin MDP, DNA, RNA, toxins |
Retinoic acid inducible gene I (Rig1)-like receptors (RLR) | DDX58 (RIG-1) DHX9, DHX36 | Short ds-RNA, ss-RNA DNA |
C-type lectin receptors (CLR) | CD207 (langerin), CD209 (DC-SIGN), CLEC6A | Fucose, mannose High mannose High mannose |
Toll-like receptors (TLR) | (cf. below) |
B) TLR
Localization | TLR subtype | Principal PAMP(s) | Mostly expressed on |
|---|---|---|---|
Extracellular | TLR1/TLR2a | Lipoproteins | Gram-positive bacteria, mycobacteria |
TLR2 | Lipoproteins, peptidoglycan (PGN) | Gram-positive bacteria | |
TLR4 | Lipoproteins, lipopolysaccharides (LPS) | Gram-negative bacteria | |
TLR6/TLR2a | e.g. lipoteichoic acid (LTA) | Gram-positive bacteria, mycoplasma | |
TLR10 | Not known | ||
TLR5 | Flagellin | Flagellated bacteria | |
Intracellular | TLR7 | Single-stranded RNA (ssRNA) | Viruses |
TLR8 | ssRNA | Viruses | |
TLR9 | CpG oligodeoxynucleotide | Bacteria; DNA viruses | |
TLR3 | Double-strain RNA (dsRNA) | Viruses |
The potency of TLR-mediated danger signals in triggering immune responses cannot be reduced to their impact on DC and other cells of hematopoietic origin. In fact, keratinocytes [17] express a series of TLR (at the mRNA level: TLR1-6 and 9-10; functionally: TLR3, 4, 5 and 9, [17, 18]). Engagement of their respective ligands can trigger (as illustrated in the following paragraphs) both innate and adaptive responses.
As far as LC and DC are concerned, studies investigating their TLR expression have yielded partially divergent results [18–21], probably due to differences in the experimental setting, e.g. culture conditions. It seems clear that LC and the various DC subsets do not share the same TLR expression patterns (cf. Fig. 1.1a, b) and, in consequence, exhibit different reactions to microbial or other immunogenic stimuli. This distinct distribution of TLR on DC allows the immune system to elegantly orchestrate innate and adaptive responses, which is why growing efforts have been put into the development of vaccine formulations making use of these mechanisms.
TLR as Gatekeepers of Tolerance Towards Bacteria?
According to the idea that LC are responsible for maintaining tolerance and DDC for initiating immune reactions, one would expect that LC do not react to the epidermal invasion of harmless, gram-positive bacteria belonging to the commensal skin flora. This theory seems to be supported by the finding that DDC abundantly secrete IL-6 and TNF-α when exposed to bacterial components (such as Pam3CSK, a synthetic TLR1/2 ligand, LPS and PGN [21]), while LC secrete IL-6, -8 and -10 only upon exposure to PGN [18, 21]. PGN-induced IL-10 could, via its inhibitory effect on the antigen presentation function of LC [22], contribute to LC-modulated tolerance towards commensal bacteria. The concept that TLR-mediated signals can contribute to maintaining tolerance is further strengthened by evidence from keratinocyte studies. The latter have shown that in keratinocytes engagement of LTA belonging to the commensal bacterium Staphylococcus (S.) epidermidis, but not to S. aureus induces an inhibitory effect on TLR3-triggered IL-6 and TNF-α expression [23] and even promotes the expression of antimicrobial peptides [24].
Orchestration of TLR-Transmitted Signals in Viral Infections
As far as viral infections are concerned, it was even before the discovery of TLR that the so-called pDC were identified as a rich source of the type I interferon IFN-alpha (IFN-α) in response to viruses (review in: [25]). IFN-α is a potent tool in the antiviral defense and acts against viruses both indirectly (by enhancing adaptive immune functions) and directly. Later, it was found that the abundant IFN-α production in pDC is triggered by signals from TLR recognizing virus components, i.e. ssRNA (TLR7) and CpG oligonucleotide (TLR9). In contrast, DDC as well as freshly isolated LC do not seem to undergo phenotypic or functional changes in response to direct exposure to these TLR ligands [18]. In the presence of CpG-stimulated keratinocytes however (which abundantly produce IL-1α, TNF-α and GM-CSF), LC up-regulate major histocompatibility complex (MHC) class II and the costimulatory molecule CD86 [26]. The complexity of TLR-transmitted “danger signals” is illustrated by the finding that dsRNA, the virus-associated ligand for TLR3, does not elicit any response in pDC (for review, cf. [25]) but instead enhances different functions in LC and DDC. In LC, the synthetic TLR3 ligand polyinosinic:polycytidylic acid (poly I:C) induces changes that promote an adaptive antiviral response. These include maturation, IL-6 production and upregulation of CD70 (a potent promoter of CD8+ T cell responses) [27]. Meanwhile, exposure of CD141+ DC to poly I:C results in IFN-γ production in CD141+ DC [11] and enhances (in a skin explant model) maturation and migration [20]. Keratinocytes respond to poly I:C by up-regulating surface molecules such as MHCII, CD40 and the Fas receptor [17] and by abundantly secreting TNF-α and IL-6 [23].
TLR-Transmitted Danger Signaling Beyond Skin Infections
A role of TLR danger signals has been demonstrated in various skin conditions beyond infections including acne vulgaris (Chap. 24), roseacea, skin cancers and psoriasis (Chap. 21) (for overview, cf. [28]). In the case of CHS, it had long been known that immunogenic haptens induce the secretion of proinflammatory cytokines in keratinocytes, LC [29] and DC and that skin inflammation is required for the development of sensitization to a hapten. The molecular events behind this remained obscure. An involvement of certain TLR in CHS was indicated by studies revealing that TLR2/TLR4 double-deficient mice are completely resistant to CHS development (see also Chap. 23). The finding that germ-free mice still develop CHS pointed towards a role of endogenous (and not necessarily microbial) ligands in eliciting inflammation during the sensitization phase. In mice, some allergens (such as 2,4,6-trinitro-1-chlorobenzene, oxazolone, and fluorescein isothiocyanate) seem to indirectly activate TLR [28]. Meanwhile, Goebeler et al. were able to demonstrate in elegant experiments that Ni2+ ions directly bind to the human TLR4 and, by doing so, initiate a signaling cascade resulting in the generation of proinflammatory signals [30]. The respective role of keratinocytes, LC and DC in TLR-mediated inflammation during the sensitization phase of CHS remains to be elucidated. The lack of TLR expression on LC for instance did not dampen CHS development in a mouse model [31].
In atopic dermatitis (see also Chap. 22), patients exhibit reduced expression of TLR2 on keratinocytes and monocytes/macrophages [32, 33]. TLR2 recognizes S. aureus-associated patterns and enhances the expression of certain tight junction molecules [34]. The deficiency of TLR2 in atopic dermatitis patients could thereby not only contribute to their susceptibility to S. aureus infections but also reinforce barrier dysfunction, a major feature of the disease.
TLR-Driven Innate Effector Functions of DC
Imiquimod: A Pharmaceutic TLR Ligand
In the early 1990s, it was reported that incubation of peripheral blood leukocytes with certain imidazoquinolines (e.g. imiquimod, resiquimod) results in the production of IFN-α by these cells. Soon, it became clear that imiquimod acts as an artificial ligand of TLR7 [35], single-strand sensing receptor important in triggering IFN-α in pDC [36].
Given the crucial role of IFN-α as a first line defense against viral infections, imiquimod has been developed into a topical cream compound (Aldara®) for the treatment of viral acanthomas such as genital warts [37]. In the years to come, Aldara® cream was also proven to be efficacious in superficial basal cell carcinomas (BCC), lentigo maligna and actinic keratoses (review in: [38]).
pDC as Effector Cells in Imiquimod-Induced Tumor Regression
We as well as other investigators set out to unravel the mode of action of topical imiquimod. In a first series of experiments, we observed that application of Aldara® cream to murine ear skin for only a few days causes massive infiltration of neutrophils, macrophages and, particularly noticeable, pDC. In subsequent experiments we transplanted a (murine) melanoma cell line into the skin of mice. After several weeks, melanomas had appeared and were then treated with either Aldara® cream or vehicle. Aldara® but not the vehicle regularly induced resolution of tumors not exceeding a volume of 130 mm3. Again, pDC were conspicuously present around and within the regressing melanoma cell islands [39]. All these findings led us to hypothesize that pDC were, in one way or the other, involved in Aldara®-induced tumor regression. In a subsequent study, we treated sporadic superficial BCC from seven patients with topical imiquimod for a total of 6 weeks and examined the clinicopathologic features of the tumor during the course of therapy [40]. After 2 weeks of treatment, BCC lesions showed signs of severe inflammation that quickly resolved after termination of therapy and left behind an area of normal-appearing skin histopathologically free of cancer cell nests (Fig. 1.2a). Immunohistological analysis of lesional skin after 2 weeks of imiquimod treatment revealed changes similar to those seen in our murine model. This was evidenced by a considerable number of apoptotic cancer cells and tumor cell islands surrounded and/or infiltrated by a dense inflammatory infiltrate that contained considerable numbers of inflammatory DC of both the myeloid and the plasmacytoid type (Fig. 1.2b, c). When we evaluated by immunohistochemistry the expression of lytic molecules, we surprisingly found granzyme B and perforin mainly on myeloid DC and TRAIL (tumor necrosis factor related apoptosis inducing ligand) mainly on pDC. Strikingly, the apoptosis-inducing TRAIL-receptor 1 was expressed on BCC (Fig. 1.2c). These in vivo data received experimental support by in vitro studies demonstrating the capacity of imiquimod to induce TRAIL on peripheral blood pDC in a strictly IFN-α-dependent manner. TRAIL-expressing, but not unstimulated pDC were perfectly capable of lysing MHCI – bearing tumor cell targets [40, 41] implying that TRAIL-positive pDC in BCC are directly responsible for the killing of the cancer cells. The presence of the pro-apoptotic TRAIL receptor 1 on BCC cells supports this notion [40] as do studies in melanoma-bearing mice treated with imiquimod [42].
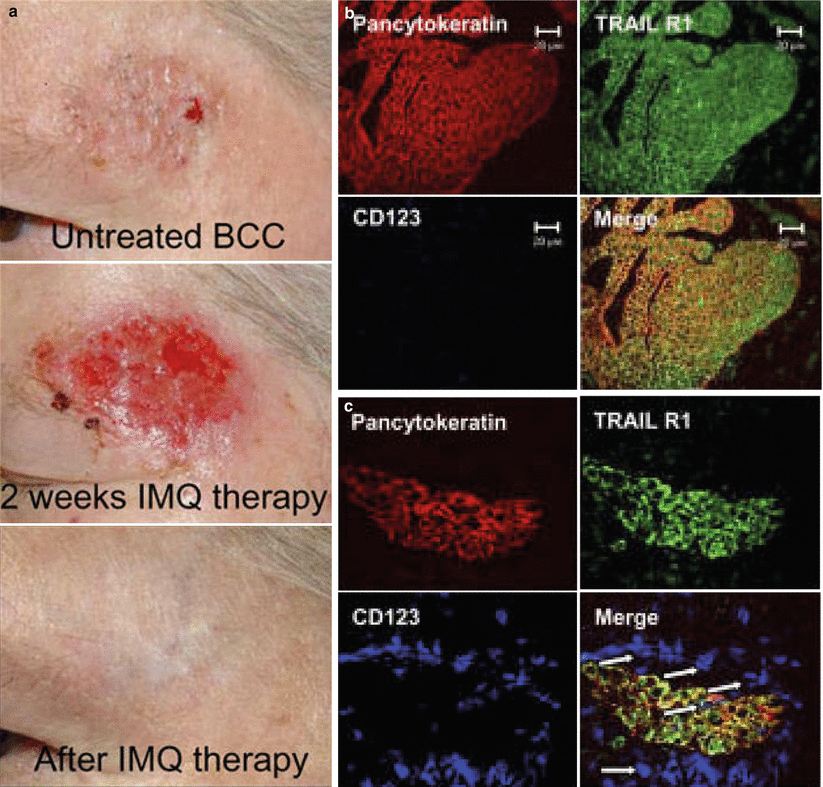
Fig. 1.2
Effects of imiquimod on BCC. (a) Imiquimod topically applied to superficial BCC five times a week for a period of 6 weeks led to a local inflammatory response, which resulted in a complete clinical and histopathological tumor clearance in all patients treated. The clinical pictures are representative for all patients (n = 7) treated with imiquimod. Immunofluorescence triple labeling of (b) untreated and (c) imiquimod-treated BCC with anti-pancytokeratin (TRITC), anti-TRAIL-R1 (A488) and anti-CD123 (Cy5) reveals TRAIL-R1+ BCC cells surrounded by CD123+ cells (arrows in c) (© 2007 Stary et al. [40])
Melanoma
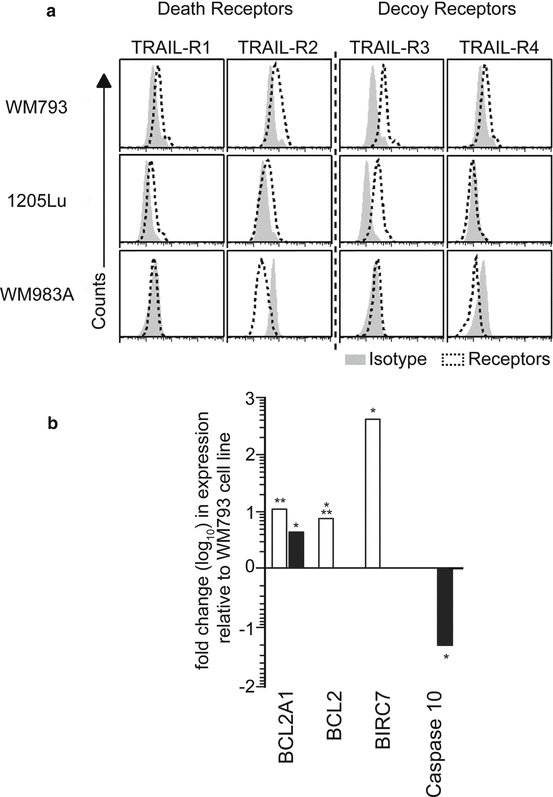
In the case of human melanoma, the situation is more complex. We have recently reported that pDC that had been rendered TRAIL-positive by imiquimod stimulation were capable of lysing certain melanoma cell lines, but not others [41]. Further investigations revealed that these differences in TRAIL sensitivity are due to distinct expression patterns of pro-apoptotic TRAIL receptors on different melanoma cell lines and, more importantly, of pro- and anti-apoptotic effector molecules within these cell lines (Fig. 1.3a, b) [43, 44]. When searching for ways to increase the TRAIL susceptibility of resistant cell lines, we found in accordance with previous reports [45] that the anti-inflammatory compound diclofenac was able to do so (Vazquez-Strauss et al., in preparation). In fact, diclofenac led to an enhanced expression of pro-apoptotic TRAIL receptors on melanoma cells as well as to an upregulation of pro-apoptotic and, vice versa, a downregulation of anti-apoptotic molecules within the cancer cells [46]. It will be interesting to explore whether the beneficial effect of diclofenac in the treatment of certain cancers is, at least partly, due to this phenomenon and, if so, whether ways can be found to maximize this tumoricidal effector mechanism.