(1)
Mill Hill Laboratory, The Francis Crick Institute, London, UK
(2)
St. John’s Institute of Dermatology, King’s College London, London, UK
Abstract
Psoriasis is a common, complex, inflammatory skin disease resulting from the interplay of genetic, environmental and immunological factors. In the last four decades, advances in understanding psoriasis etiopathogenesis have resulted from pivotal genetics studies, as well as, the integration of clinical and experimental models of disease. This has resulted in the identification of several psoriasis susceptibility genes as well as cellular and molecular mediators, with some of these findings already translated in novel targeted therapies. Here we review the psoriasis literature, describing the elucidation of key pathogenic mechanisms and their translation into effective drugs. Moreover, we describe how the stratified medicine approaches being developed in psoriasis and the quest for psoriasis biomarkers are poised to predict individual susceptibility to disease, detect the onset of disease at the very earliest stages pre-empting its progression, and to develop and prescribe safe and effective medicines to each patient.
Keywords
SkinGeneticsT cellsKeratinocytesTargeted therapiesBiomarkerStratified medicineIntroduction
Psoriasis is a complex disease, resulting from the interaction of genetic, environmental, and immunological factors [1, 2]. The complexity of its etiopathogenesis is mirrored by a spectrum of clinical phenotypes, which often associates with co-morbidities that are also multi-factorial and possibly feed back onto the primary disease. Being one of the most common skin conditions, psoriasis has received a great deal of attention from clinicians and basic scientists alike in the past four decades, becoming a model to study chronic skin inflammation. This joint effort has resulted in the elucidation of many underlying pathogenic mechanisms and, more importantly, has been translated in novel therapeutic strategies. Here, we review recent advances in understanding the complex genetic, environmental and immunological basis of psoriasis, identify research gaps which need to be filled in, describe how some of the more recent findings have resulted in novel targeted therapies already available in the clinic or in advanced stages of clinical trials, and highlight how stratified medicine approaches and the quest for psoriasis biomarkers has begun in earnest to further improve patients’ life.
Epidemiology, Clinical Subtypes and Histological Features
Psoriasis is a common disease, affecting 2–4 % of the population in western countries, with prevalence rates influenced by age, geographic location and genetic background [3]. Prevalence is higher in adults (from 0.91 to 8.5 %) as compared to children (from 0 to 2.1 %) with a dual peak of incidence: an early onset, before the age of 40, which is often associated with familiar disease history and showing high association with the human leukocyte antigen (HLA)-Cw0602 allele (type I psoriasis), and a late onset, after the age of 40 (type II psoriasis). Geographical patterns of prevalence suggest lower prevalence in countries closer to the equator, in keeping with the beneficial effects of UV radiation exposure [4]. Prevalence is higher in individuals of European descent (from 0.73 to 2.9 %) as compared to those of African and Asiatic background (from 0 to <0.5 %).
Psoriasis has traditionally been considered to affect both genders equally; however recent data about age stratification within gender shows a higher incidence in females <18 years old, and conversely a higher incidence in males ≥18 years old [5, 6].
The term psoriasis (from the Greek word psora, to itch) encompasses a number of distinct clinical phenotypes [7]. This heterogeneity often represents a dynamic, anatomical or qualitative spectrum of the same disease (e.g. large and small plaque-psoriasis), while in other cases most likely pins down the existence of quite different disease entities (e.g. generalized pustular psoriasis, GPP). According to the International Psoriasis Council, there are four main forms of psoriasis: plaque-type, guttate, GPP, erythroderma, plus several further sub-phenotypes defined based on different parameters; i.e. the distribution, anatomical localization, size and thickness of plaques, onset and disease activity [8].
Plaque-type psoriasis, occurring in 85–90 % of affected patients is the most common form of psoriasis, the most studied and also the one usually targeted in randomized controlled clinical trials. Plaque-type psoriasis is characterized by oval- or irregularly-shaped, red, sharply demarcated, raised plaques, covered by silvery scales [1, 7] (Fig. 21.1). Plaques occur mainly on the extensor surface of elbows and knees, on the scalp and in the lower back, but can affect every area of the body, often with a symmetrical distribution. Key histological features of psoriasis are epidermal thickening (acanthosis), incomplete keratinocyte terminal differentiation with retention of the nucleus by corneocytes (parakeratosis) and thickening of the statum corneum (hyperkeratosis), elongation of the rete ridges extending downward between dermal papillae (papillomatosis), together with blood vessel dilation and immune cell infiltration into the skin. In the epidermis, neutrophils accumulate into the parakeratotic scales in the stratum corneum forming Munro microabscesses while lymphocytes, mainly CD8 + T cells, are interspersed between keratinocytes. The dermis is heavily infiltrated by T cells (mainly CD4+) and dendritic cells (DC).

Fig. 21.1
Clinical and histoimmunological features of psoriasis- (a–c) clinical photographs of chronic plaque-psoriasis, note nail involvement in (b). (d) Hematoxylin-stained skin section from chronic plaque-psoriasis showing acanthosis, papillomatosis, parakeratosis, as well as Munro abscess. (e) Immunofluorescence staining of chronic plaque-psoriasis showing skin-infiltrating CD3+ T cells in green (Reproduced with permission from: Di Meglio et al. [242])
Psoriasis is a dynamic disease: newly formed lesions evolve into an advanced plaque that can slowly enlarge (active lesion) or remain static (stable lesion) [8]. Resolving lesions after therapy can be encased by a distinctive rim of blanching (Woronoff’s ring), predictive of clearing, and are histologically characterized by orthokeratosis, that is thickening of the stratum corneum without parakeratosis and restoration of the stratum granulosum.
Guttate psoriasis, from the Latin “gutta” for tear drop, is characterized by multiple small scaly plaques, commonly occurring around the trunk and upper arms and thighs. The rash has often abrupt onset, usually within 2–4 weeks after streptococcal pharyngitis in children and young adults and is therefore associated with type I psoriasis [8]. Guttate psoriasis can either completely clear spontaneously or following topical treatment, become chronic, or worsen into the plaque-type.
Generalized pustular psoriasis (GPP), also known as von Zumbush psoriasis, is a rare but potentially life-threatening disease characterized by episodic, widespread skin and systemic inflammation. A typical histological feature of GPP is the presence of conspicuous aggregates of neutrophils infiltrating the stratum spinosum (spongiform pustules of Kogoj) and giving rise to sterile cutaneous pustules [7]. Acute attacks can occur during pregnancy and may be triggered by infection, exposure to or withdrawal of drugs. GPP is frequently associated with plaque type psoriasis and/or palmoplantar pustular psoriasis (PPP). Although still classified as a variant of psoriasis, the salient clinical and histological feature of GPP have long suggested that is a disease of distinct aetiology.
Recent genetic data lend further support to this hypothesis suggesting that, at least in certain cases, GPP is inherited as an autosomal recessive trait, due to mutations in the IL36RN gene encoding the anti-inflammatory IL-36-receptor antagonist, IL-36Ra [9, 10]. IL-36Ra blocks the pro-inflammatory cytokines IL-36α/β/γ: when IL36RN is mutated, IL-36 signaling is unrestrained, with enhanced production of further pro-inflammatory cytokines [10]. However, IL36RN mutations only occur in a minority of patients [11], thus more genetic determinants are likely to be involved. Interestingly, a de novo mutation in the epidermal NF-kB activator CARD14 [12] has been described to underlay a sporadic case of severe GPP, suggesting that keratinocytes dysfunction is likely to play a predominant role in this disease phenotype.
Erythrodermic psoriasis is characterized by diffuse erythema, with or without scaling, involving more than 75 % of the skin surface. It represents the most severe, albeit rare, psoriasis phenotype. Systemic manifestations such as hypothermia and limb oedema might occur, due to the generalized vasodilation underlying the erythema, as well as myalgia, fatigue and fever. Both administration and abrupt withdrawals of systemic corticosteroids or methotrexate, sunburn and emotional stress have been suggested as possible triggering factors [13].
Added together, these last three forms of psoriasis represent 10–15 % of psoriasis cases. Both their relative infrequency as compared to the plaque-type form, and their peculiar epidemiological and clinical features (e.g. high prevalence in children and young adults for the guttate form or the extremely severe systemic manifestation for GPP and erythrodermic psoriasis) have hampered so far a thorough investigation of the genetic determinants and molecular and cellular mechanisms underlying their pathogenesis, which might diverge from that of plaque-psoriasis. Moreover, very few or no randomized controlled clinical studies have been specifically conducted with these forms of psoriasis thus far, leaving open only the option for off label use of approved therapy for plaque-type psoriasis which might or might not be effective. Thus, a concerted effort by the wide international scientific community is requested to fill both these gaps in the near future.
Co-morbidities
Psoriatic Arthritis
About 20–30 % of psoriasis patients develop psoriatic arthritis (PsA), a seronegative, chronic inflammatory muscoskeletal disorder, occurring in most cases about a decade after the appearance of psoriasis [14].
PsA has a complex aetiology mirrored in a wide spectrum of clinical disease presentation, expression and clinical course [15]. PsA can affect different tissues (synovium, cartilage, bone, entheses, tendons); it presents common involvement of distal joints, asymmetric articular distribution, erythema over affected joints, spinal involvement and enthesitis [14] and eventually leads to erosion and loss of function of the affected areas.
Since about 80 % of the patients develop PsA following psoriasis [16], PsA is sometimes considered as a disease within a disease with the skin manifestation being the parent disease [17]. PsA has a stronger genetic component than psoriasis (sibling recurrence risk (λs) of 27–47 for PsA v [18] vs. 4–11 for Ps [19]), however it is not as well defined as that of psoriasis due to phenotypic heterogeneity, disease overlap and smaller number of patients analyzed. Several PsA susceptibility genes, such as HLA–C, IL12B, IL23R, TNIP1, overlap with psoriasis [16, 20, 21]. On the other hand, differences in the genetic background of the two conditions do exist and unique genetic determinants have been identified, although not at a genome-wide significant level [21]. Nevertheless, PsA shares several key cellular and molecular mediators with psoriasis, such as lymphocytes infiltrating the inflamed skin or joint [22] and the pro-inflammatory cytokines TNF, IL-23, and IL17. TNF is a critical disease player and about 70 % patients successfully respond to anti-TNF therapy in terms of signs and symptoms improvement, and, in some cases also by radiographic progression [15]. Moreover, clinical trials showed efficacy for the anti IL-12/IL23p40 antibody ustekinumab [23–25] or anti-IL17 secukinumab [26] in the treatment of PsA.
Other Co-morbidities
The association of psoriasis with physical and psycho-social co-morbidities, such as cardiovascular disease (CVD, i.e. myocardial infarction (MI) and stroke), metabolic disorders (obesity, non-alcoholic fatty liver disease, dyslipidaemia and diabetes), depression and Crohn’s disease (CD), is increasingly being appreciated and results in a more modern definition of the disease as a systemic inflammatory disorder in which the chronic nature of the skin inflammation is likely to contribute to the development of associated co- morbidities [27]. In analogy with that occurring in atopic dermatitis, the concept of “psoriatic march” has been proposed to describe the synergistic interplay of psoriasis and its co-morbidities in the establishment of systemic inflammation [28].
Moreover, in analogy to hypertension being called the “silent killer”, psoriasis has been recently dubbed the “visible killer” [29] as patients with severe psoriasis have a 6-year reduction in life expectancy, mainly due to excess risk of CV death [30, 31]. The association between psoriasis and CVD has attracted considerable interest since a significant increased risk of death from CVD for psoriasis patients requiring hospital admission was reported in 2004 [32]. Nevertheless, it has been long known that psoriasis patients have a higher prevalence of traditional cardiovascular risk factors such as diabetes, hypertension, dyslipidaemia, obesity, and metabolic syndrome compared to the general population [33–35] that might account for some or all of the increased CVD risk. Therefore, a number of population-based epidemiological studies have been performed in the last decade to address this issue. Unfortunately, these studies are often heterogeneous in their data collection methods, outcomes, sample size and thus in statistical power, and display variable degrees of control for confounding factors [29, 36, 37]. This heterogeneity is likely to account for some conflicting results [38–40].
Nevertheless, a growing body of epidemiologic literature suggests that severe psoriasis (defined in most of the studies as affecting >10 % body surface area or requiring systemic treatment or phototherapy) confers a clinically significantly increased risk of CVD and cardiovascular mortality that is independent of conventional risk factors [39, 41]. A recent meta-analysis, including 14 previous epidemiological studies of which ten were population-based, showed increased risk of CVD in patients with severe disease, with OR relative to the general population of 1.37 for CVD mortality, 3.04 for myocardial infarction (MI) and 1.59 for stroke [42]. Thus, psoriasis has been included as an independent risk factor for CVD in recent guidelines for CVD prevention (Fifth Joint Task Force of the European Society of Cardiology, 2012).
Moreover, severe psoriasis has been shown to be an independent risk factor for atherosclerotic CV disease, as defined by outcomes such as imaging of coronary and carotid arteries, and measurements of endothelial function and arterial stiffness [43–45].
As mentioned earlier, high prevalence of metabolic syndrome has been reported among psoriasis patients, with more than two-fold increased odds ratio (OR) in psoriasis patients, compared to matched healthy controls [46]. In particular, the association of type 2 diabetes with psoriasis is stronger in patients with severe disease (OR = 1.97) as compared to those with mild disease (OR = 1.53).
Clinical data and a better understanding of psoriasis immunopathogenesis also support these epidemiological observations. Elevated levels of nonspecific inflammation markers (e.g. as C-reactive protein), pro-inflammatory cytokines (such as TNF and IFN-γ) and immune cells (such as T helper type 1 (Th1) and Th17) in the circulation of psoriasis patients [47, 48], and the fact that most of these inflammatory markers are also increased in the skin lesions [49], strongly support that psoriasis is not only skin deep. Indeed, psoriasis patients have high levels of lipids and lipid peroxidation, altered adipokine function [50, 51] as well as abnormal coagulation profile [52, 53].
Despite a definitive biological link between co-morbidities and psoriasis has yet to be identified, it is reasonable to infer that the systemic inflammation and dyslipidaemia present in patients may predispose to impaired glucose tolerance and cardiovascular damage [27]. It has been suggested that the pro-inflammatory molecules produced by the skin could be released into the systemic circulation; in fact several genes differentially regulated in psoriasis are linked to functional pathways associated with metabolic diseases/diabetes and cardiovascular risk [49]. For instance renin, an enzyme involved in the renin–angiotensin pathway ultimately regulating blood pressure, is over-expressed in psoriatic skin, suggesting a functional link between expression profile at the skin level and peripheral functions. The presence of IL-17A/IL-17 F and CD4+ cells expressing IL-17 and IFN-γ in atherosclerotic lesions [54, 55], and risk alleles shared between psoriasis and its metabolic and cardiovascular co-morbidities [56], also lend support to the association between psoriasis and these diseases.
Epidemiological [57–59] and genetic [60, 61] studies support a close relationship between psoriasis and inflammatory bowel diseases (IBDs). Increased occurrence of psoriasis has been reported in patients with CD (8.9 %) yielding an aggregate relative risk of 7.1 (χ 2 = 139, P < 1 × 10−9) [62]. Conversely, Li et al recently reported that women with psoriasis have a significantly increased risk of CD (relative risk (RR), 3.86, 95 % CI 2.23–6.67), but not ulcerative colitis (UC) (RR, 1.17, 95 % CI 0.41–3.36) [58].
Remarkably, psoriasis and IBDs share a number of common genetic determinants such as IL23R [60, 61].
Finally, psoriasis carries a severe psychosocial burden with anxiety, depression and perceived stress appearing at higher rate in psoriasis patients [63]. The impact of psoriasis on health-related quality of life is similar to that of other major medical diseases, including cancer, arthritis, hypertension, heart disease, diabetes, and depression [64]. Psoriasis patients find it hard to adapt to the chronic yet variable and unpredictable nature of the disease. Another major component of psychological distress is the anticipated negative reactions of others which can be of shame or stigmatization. Coping mechanisms include avoidance and seclusion which in turn affect their quality of life.
Changes in cognitive processing of facial expressions of disgust have been identified in psoriasis patients, who display smaller signal responses to disgusted faces to protect them from stressful emotional responses [65].
Depression, which is one of the stronger predictors of suicidal ideation, has been observed in more than 60 % of patients [66], and higher prevalence of suicidal ideation has been detected in psoriasis patients as compared to healthy controls and patients affected by other skin disease [67, 68].
Taken together, clinicians are presented with the challenging task of managing a multifaceted and lifelong disease which, although apparently not lethal, can severely affect patients’ quality of life and, in some cases, life expectancy.
Thus, the current focus is not to only treat but to manage psoriasis patients [69]. A multidisciplinary approach is needed, particularly in cases of severe psoriasis with multiple comorbidities or cardiovascular disease with dermatologists alerted to detect and react to early indications of cardiovascular risk factors and comorbidities in psoriasis patients. While all patients should be encouraged to correct their modifiable cardiovascular risk factors, particularly obesity and smoking, and to adopt healthy lifestyle behaviours, patients with moderate to severe psoriasis are to be recognized and managed as being at intermediate risk of CVD with appropriate counselling and treatment [70].
Moreover, a patient-centred therapeutic approach, undertaken early in the psoriasis treatment pathway (“early intervention”) aimed at complete clearance, has been advocated to improve control of cutaneous symptoms and modify disease course and burden [71].
Etiopathogenesis
Psoriasis is a complex disease occurring in genetically predisposed individuals, in which a dysregulated immune response takes place following exposure to certain environmental triggers.
Genetics
Genetic predisposition to psoriasis is supported by population and family studies, as well as, higher concordance rates in monozygotic twins, compared with dizygotic twins (up to 73 vs. 20 %, depending on the population studied) [73–76]. Lack of complete concordance between monozygotic twins and familial occurrence of disease not following a clear inheritance pattern, support the definition of psoriasis as a complex genetic trait, resulting from gene–gene and gene–environment interactions [77, 78]. Large efforts to understand the genetic architecture of psoriasis have resulted in the identification of a number of psoriasis genetic determinants. The psoriasis genetic landscape emerging from recent genome-wide association studies (GWAS) and their meta-analysis [20, 60, 79–86], as of mid-2014, includes 36 independent psoriasis-associated genetic regions in individuals of European ancestry (Table 21.1), plus five more uniquely associated in the Chinese population [87]. Psoriasis- susceptibility genes encompass skin and immune-related genes, with the latter belonging to either the innate or the adaptive immunity, as well as bridging the two arms of the immune system. SNPs and Copy Number Variation (CNV) [85, 86] in genes of the late cornified envelope (LCE) family within the epidermal cell differentiation complex, a cluster of genes involved in skin barrier formation, support a critical role for skin-specific genes in psoriasis susceptibility. Among immune genes, the over-representation of four pivotal immunological processes and pathways strongly points towards their critical contribution to disease susceptibility: antigen presentation (HLA–C and ERAP1), NF-kb signalling (e.g. TNFAIP3, TNIP1, TRAF3IP2, CARD14), type I IFN pathway (e.g. IL28RA and RNF114), and IL-23/IL-17 axis (e.g. IL23A, IL12B, and IL23R) [77]. Psoriasis susceptibility region 1 (PSORS1) within the major histocompatibility complex is the strongest susceptibility locus [88, 89] and the HLA- Cw*0602 allele of the MHC class I molecule HLA-C is considered to be the primary associated allele, as confirmed by early sequence and haplotype analysis [90], and more recently by GWASs [81, 82, 86], and analysis of high density SNP data [91]. The HLA-C association has not only the greatest statistical significance observed in GWAS studies [86] but also accounts for about 6 % of the total genetic variance [92], which is at least ten fold more than that explained by any other known psoriasis susceptibility locus [86]. The expression pattern of MHC class I molecule on all nucleated cells makes HLA-C capable of regulating both innate and adaptive responses [93]. Nevertheless, despite the strong genetic evidences and the obvious immunological function of HLA-C, functional studies addressing the precise mechanism by which -Cw*0602 alleles predispose to psoriasis are still missing and no -Cw*0602 specific antigen or interacting protein has been identified to date. Among genes of the innate immunity, more than half belong to the NF-kB pathway, which has a pivotal role in amplifying and sustaining chronic inflammation. Two recent studies have identified and evaluated the functional consequences of rare and common gene variants [86, 94] and missense mutations [12] in CARD14, an activator of NF-kB primarily expressed in skin epidermis. Genes belonging to the type I IFN pathway support clinical and experimental findings indicating an important role for antiviral responses in psoriasis [95, 96]. Finally, several susceptibility genes belong to the IL-23/IL17 pathway, whose critical involvement in disease pathogenesis has been extensively documented by a wealth of studies showing a pivotal role for IL-23-induced and IL-17-mediated responses in psoriasis [97]. Moreover, the genetic association with IL23R is one of the very few supported by functional evidence with reduced IL-17 responses in carriers of the protective Arg381Gln IL23R allele [98, 99]. Interestingly, some genes from the above pathways influence multiple phenotypic traits, in particular other immune-mediated conditions such as Crohn’s disease (CD), celiac disease and ankylosing spondylitis [86], thus confirming the presence of a shared genetic basis among immune-mediated inflammatory diseases.
Table 21.1
Psoriasis susceptibility genes
Chr | Gene(s) | Class | Protein function | Pathway | Reference |
|---|---|---|---|---|---|
1 | LCE3B/3C/3D | Skin-related | Keratinocyte structural protein | Skin barrier formation | |
1 | IL28RA | Immunity | IL-29 receptor subunit | IFN signaling | [82] |
1 | IL23R | Immunity | Unique subunit of IL-23 receptor complex | IL-23/IL17 axis | |
1 | RUNX3 | Immunity | Transcription factor | Tbet pathway | [86] |
1 | TNFRSF9* | Immunity | Adaptor molecules involved in T cell biology | T cell differentiation | |
2 | REL | Immunity | NF-kB subunit | NF-kb signaling | [82] |
2 | IFIH1 | Immunity | Innate antiviral receptor | IFN signaling | [82] |
2 | B3GNT2 | Other | Enzyme | Carbohydrate metabolism | [86] |
5 | TNIP1 | Immunity | Inhibitor of TNF-induced NF-kB activation | NF-kb signaling | |
5 | IL12B | Immunity | Shared subunit of IL-12/IL-23 | IL-23/IL17 axis | |
5 | ERAP1 | Immunity | Enzyme processing MHC class I ligands | Antigen presentation | |
5 | IL4/IL13 | Immunity | IL-4 and IL-13 cytokines | IL-4/IL-13signaling | [81] |
6 | TNFAIP3 | Immunity | Inhibitor of TNF-induced NF-kB activation | NF-kb signaling | |
6 | TRAF3IP3 | Immunity | Adaptor molecule mediating IL-17-induced NF-kb activation | IL-23/IL17 axis, NF-kB signaling | |
6 | IRF4* | Immunity | Transcription factor | IL 17 signaling | [86] |
6 | HLA-C | Immunity | MHC class I antigen | Antigen presentation | |
6 | TAGAP | Immunity | RhoGTPase-activating protein | T cell activation | [86] |
7 | ELMO1 | Immunity | Involved in TLR-mediated IFN-a signaling | IFN signaling | [86] |
9 | KLF4 | Skin-related/immune | Transcription factor | Skin barrier formation, IL 17 signaling | [86] |
9 | DDX58 | Immunity | Innate antiviral receptor | IFN signaling | [86] |
10 | ZMIZ1 | Immunity | Protein inhibitor of activated STAT(PIAS) family of proteins | TGF-β signaling | |
11 | PRDX5 | Other | Antioxidant enzyme | Intracellular redox signaling | |
11 | ETS1 | Immunity | Transcription factor | Unknown | [86] |
11 | ZC3H12C | Other | Zinc finger protein with putative RNase function | Unknown | [86] |
12 | IL23A | Immunity | Unique subunit of IL-23 | IL-23/IL17 axis | |
14 | NFKBIA | Immunity | Inhibitor of NF-kB activation | NF-kb signaling | |
16 | FBXL19 | Immunity | Putative inhibitor of NF-kB activation | NF-kb signaling | [83] |
16 | SOCS1 | Immunity | Suppressor of cytokine signaling | Type II IFN signaling | [86] |
17 | CARD14 | Immunity | Activator of NF-kB pathway | NF-kb signaling | |
17 | NOS2 | Immunity | Induced nitric oxide synthase | Inflammation | [83] |
17 | STAT3* | Transcription factor | IL-23/IL17 axis | [86] | |
18 | MBD2* | Other | Transcriptional repressor | Unknown | [86] |
19 | CARM1* | Immunity | Transcriptional co-activator of NF-kB | NF-kb signaling | [86] |
19 | TYK2 | Immunity | Tyrosine kinase associated with cytokines receptors | IL-23/IL17 axis, IFN signaling | [82] |
20 | RNF114 | Immunity | E3 ubiquitin ligase | IFN signaling | |
22 | UBE2L3* | Immunity | Ubiquitin conjugating enzyme | NFkb signaling |
The signals identified in the European population collectively account for approximately 20 % of estimated psoriasis heritability, and the gene variants identified have only modest- effect size [86]. It has been hypothesized that rare variants with bigger effects may explain this “missing heritability” [100]. However, two recent re-sequencing studies in individuals of European or Chinese descent, have shown that rare variants at known immune-related loci have a negligible role in psoriasis genetic susceptibility [101, 102], suggesting that the estimated missing heritability either results from the co-existence of many common variants of weak effect, or has been overestimated, owing to the existence of gene-gene and gene-environment interactions [103].
Environmental Factors
In contrast to the fast-growing list of psoriasis susceptibility genes, the environmental component concurring in initiating the disease is still ill-defined. Among known environmental triggers of psoriasis there are drugs, infections, physical trauma, smoking, alcohol and stress.
Drugs, such as the anti-viral and anti-proliferative agent imiquimod, anti-depressants (lithium), anti-hypertensives (beta-blockers), cytokines (IFN-α) and anti-cytokine therapies (anti-TNF) have all been clinically associated with initiation, exacerbation and worsening of psoriasis [104]. Imiquimod, a Toll Like Receptor (TLR) 7/8 agonist used to treat genital warts and non-melanoma skin tumours, represents one of the best investigated examples of psoriasis triggers so far. The initial clinical observation of a case of psoriasis exacerbated by topical treatment with imiquimod [105] has prompted research into plasmacytoid DC (pDC) and type I IFN pathway, which is downstream of TLR7/8 signaling [95]. Moreover, it has also been translated into the imiquimod-induced psoriasiform skin inflammation mouse model, which faithfully reproduces most of the features of the human disease and has quickly become one of the most widely used experimental models to study psoriasis [106].
An association between preceding streptococcal throat infection and psoriasis [107] has been reported, mainly with guttate psoriasis [108] and homologous T cell clones have been found in both the tonsils and skin lesions of plaque-type psoriasis patients [109].
Tattoos and surgical incisions give rise to the Koebner phenomenon with psoriasis plaques appearing at the site of the trauma [110].
The association of modifiable behavioural risk factors, such as smoking and alcohol consumption, as well as co-morbidites such as stress, are traditionally more difficult to investigate. Although a number of studies have offered evidence linking stress and smoking with psoriasis [33, 111, 112] there is no consensus on whether these factors do actually cause or aggravate psoriasis [113].
While more often included among risk factors, environmental cues can also have a protective role against disease. We have recently uncovered an unexpected protective role in psoriasis for the ligand-activated transcription factor Aryl hydrocarbon receptor (AhR) [114], an environmental sensor which responds to a wide range of stimuli including environmental pollutants (e.g. dioxin), but also more physiological ones (e.g. tryptophan metabolites of dietary or light-exposure origin), by inducing detoxifying enzymes belonging to the cytochrome P450 family. In particular, physiological ligands are believed to mediate the beneficial effect exerted by AhR, via short-term activation of the receptor in contrast to the prolonged and deleterious activation sustained by dioxin and tobacco smoke [115]. By combining data from the imiquimod model of psoriasis-like skin inflammation in mice lacking AHR, with the analysis of human psoriasis skin biopsies treated ex vivo with AhR ligands we found that absence of AhR signalling results in an exacerbation of psoriasis [114]. In particular AhR exerts its beneficial effect in keratinocytes as in its absence they become over-reactive to pro-inflammatory stimuli and release a greater amount of cytokines and chemokines, thus instigating an excessive inflammatory reaction. Thus, physiological AhR activation acts as an immunological brake that prevents dysregulation of the inflammatory response. Based on this data it is tempting to speculate that psoriasis patients might have impaired activation of the AhR pathway, possibly due to genetic variants in genes of the AhR pathway or perhaps shortage of physiological ligands, and ongoing studies are addressing these hypotheses.
Immunopathogenesis
The contribution of the immune system to psoriasis is not less complex than the overall disease pathogenesis, with a variety of innate and adaptive immune cells and pro- inflammatory mediators involved, possibly at different stages of the disease.
The recent integration of findings drawn from studies of clinical samples, xenotransplant models in which human skin is transplanted onto immuno-compromised hosts, and experimental mouse model of psoriasis-like skin inflammation, has contributed and accelerated a better understanding of disease pathogenesis. It is worth highlighting that, although no mouse model can fully recapitulate the development and features of a disease only occurring in humans [116], lessons from a number of animal models, exhibiting most of the crucial clinical traits and molecular signatures of psoriasis [117], should not be discarded as they can provide valuable insights to dissect pathogenic mechanisms.
The question of whether psoriasis is primarily an epithelial- or immune-mediated disease has recurred for several decades in the scientific community, with researchers torn between the prominent changes in the skin and the increasingly recognized importance of immunological pathways. Not surprisingly, given the macroscopic alterations occurring in psoriatic skin, the focus has initially been on keratinocytes and the accelerated and aberrant terminal differentiation program they undertake in psoriasis. Following the unexpected efficacy of serendipitously administered immunosuppressive agents, immune cells have attracted attention and have been almost the exclusive focus of clinical and experimental studies for two decades. These efforts have elucidated many of the pathogenic immune mechanisms and highlighted the critical contribution of tissue resident T cells and TNF, leading to effective anti-T cell and anti-cytokine targeted therapies. Nevertheless, KC, equipped with innate immune receptors and actively taking part in inflammatory skin responses, are nowadays considered non-hematopoietic immune cells and gained the status of skin sentinel cells [118, 119], thus prompting a re-evaluation of their role in psoriasis.
The current view of psoriasis pathogenesis implies that the aberrant immune and epidermal response seen in psoriasis is sustained by a pathogenic cross-talk between epithelial and immune cells [120, 121]. This interplay is primarily driven by the critical pro-inflammatory molecules, TNF, IL-23 and IL-17, whose direct therapeutic targeting has proven to be clinically effective, with other mediators, such as IFN-α, IFN-γ and IL-22 possibly contributing to the initiation, amplification and maintenance of the disease.
One of the best characterized initiation mechanisms, leading to dysregulated skin immune responses, involves damaged skin KC releasing the cationic antimicrobial peptide (AMP) LL-37, following physical trauma or infection. LL37 binds to self-DNA/RNA fragments forming LL-37/self-DNA/RNA complexes [122–124] that activate TLR7/9-bearing pDC. pDC are a subset of circulatory DC normally absent in human healthy skin but highly infiltrating developing psoriasis lesions [95], and once activated, specialize in releasing type I IFN which is thought to play important roles in the early phases of psoriasis development. In the AGR129 xenotransplantation model, where human non-lesional psoriatic skin is transplanted onto AGR129 mice lacking T/B cells and having severely impaired NK activity, Type I IFN triggers activation and expansion of autoimmune T cells present in the transplant, leading to full-fledged psoriasis plaque formation [95]. Moreover, self-RNA-LL-37 complexes, and pro-inflammatory cytokines IL 1β, IL 6, TNF and IFNα activate myeloid DC. Psoriatic skin lesions have a 30-fold increase in myeloid dermal DC (DDC) [125] and harbour both “classical” CD11c + CD1c + DDC, also found in healthy skin, and a distinct population of “inflammatory” CD1c- DC [126] producing TNF, iNOS, IL-20 and IL-23 [127–129]. DDC are thought to migrate to skin-draining lymph nodes to present an as-yet elusive antigen (either of self or of microbial origin) to naïve T cells. T cells, particularly those residing in the skin as tissue-resident memory T (TRM) cells, are critical players in the initiation phase of disease. TRM are memory cells which do not circulate but are strategically positioned as the first-line defence in the tissue [130, 131] and have been implicated in long term peripheral immunity [132, 133]. One of the very first evidences of their existence came from the AGR xeno- transplantion model of human non-lesional psoriatic skin onto immunodeficient mice. In this model development of full-fledged psoriatic lesions occurs in the absence of T-cell recruitment from blood [134] and depends on the ability of locally activated skin TRM cells, present in the initial graft, to migrate into the epidermis [135].
Activation of DDC and their interaction with T cells is central to plaque progression as it creates an IL-23/IL-17 inflammatory environment in which DC and macrophages-derived IL-23 promotes an IL-17-rich pro-inflammatory environment sustained by Th17 cells, [48, 121], Tc17 [136–138], γδ-T cells [139, 140], NCR+ group 3 Innate lymphocytes (ILC3) [141–143] and possibly neutrophils, mast cells [122] and regulatory T cells [144]. The initial definition of psoriasis as Th1 and IFN-γ-driven disease, based on a strong type II IFN transcriptomic signature and the high frequency of Th1 and Tc1 cells in both psoriasis plaques and peripheral blood [145–147], has been challenged by the discovery of IL-23 and Th17 cells and a wealth of genetic, clinical and experimental findings indicating a key role for the IL-23/IL-17 axis in psoriasis [97].
IL-17A and IL-17 F, sharing high structural and functional homology, activate keratinocytes to produce an array of molecules with chemoattractant properties, including neutrophil- (CXCL1, CXCL2, CXCL5, CXCL8) and T cell- (CCL20) recruiting chemokines and AMP (LL37, S100A7/8/9/15) [148, 149]. Moreover, another IL-17 family member, IL-17C induces an autocrine pro-inflammatory loop in KC [150, 151] and IL-22, produced by Th, Tc and NCR+ ILC3, which mediates most of the epidermal hyperplasia by impairing KC differentiation [152, 153]. Finally, a recent study has identified IL-9-producing Th cells FOXP3+ in psoriatic lesions although their pathogenic relevance has not been established to date [154].
Taken together, a pathogenic cross-talk between KC, DCs and T cells, sustained by TNF, IL-23 and IL-17 and supported by other immune cell players and further pro-inflammatory molecules, underlie the dysregulated immune response observed in psoriasis (Fig. 21.2).
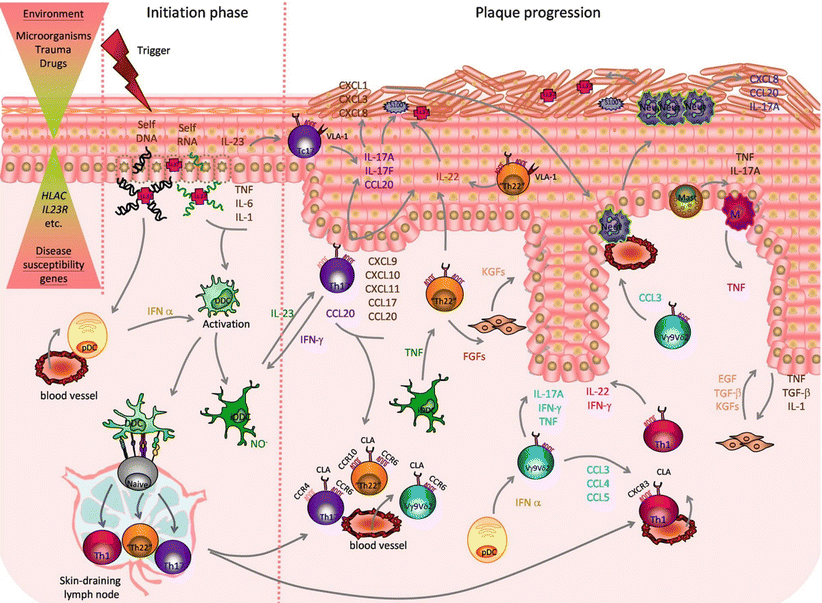
Fig. 21.2
Psoriasis etiopathogenesis- The combination of environmental factors with psoriasis susceptibility genes triggers a cascade of pathogenic events leading to disease initiation and plaque formation. In the initiation phase, pro-inflammatory cross-talk between damaged keratinocytes (KCs), releasing self-nucleic acids and LL-37, recruited plasmacytoid dendritic cells (pDCs), activated dermal DC (DDCs) and inflammatory DDC (iDDCs), producing IL-23, TNF and nitric oxide radicals (NO.), promote the activation of skin resident and newly recruited T cells that lead to plaque formation. IL-23 stimulates T helper 17 (Th17) and T cytotoxic 17 (Tc17) cells, expressing cutaneous leukocyte antigen (CLA), CCR6 and CCR4, plus very late antigen (VLA)-1 in the epidermis, to release IL-17A, IL-17 F, IL-22 and IFN-γ. IL-17A and IL-17 F act on KCs promoting production of T cells and neutrophils-attracting chemokines (CXCL1,3,8-11;CCL17-20) and antimicrobial peptides (AMPs): S100 proteins and LL-37. IL-22, produced by Th17 cells, as well as Th1 cells, expressing CXCR3 and skin-homing marker CLA, and “Th22”/“Tc22” cells, expressing CCR6, CCR10 and CLA, induces epidermal hyperplasia by impairing KC terminal differentiation. Recruited unconventional V 9v 2 T cells, expressing CLA and CCR6, are activated by pDCs-derived IFN-and release further pro-inflammatory cytokines and chemokines (CCL3-5) attracting neutrophils (Neut) and Th-1. Infiltrating Neut, mast cells and macrophages (M) contribute to the pro-inflammatory environment producing cytokines (IL-17A, TNF), AMPs (S100 proteins, LL-37) and chemokines. Cross-talk between keratinocytes producing IL-1, TNF and transforming growth factor beta (TGFβ), and fibroblasts, which in turn release keratinocyte growth factor (KGF), epidermal growth factor (EGF) and TGFβ, and possibly Th22 cells releasing fibroblast growth factor (FGFs), contribute to tissue reorganization (Reproduced with permission from Di Meglio et al. [120])
Targeted Therapy
No definitive cure is available for psoriasis and current treatments are aimed at decreasing disease activity and improving symptoms. Therapies are administered according to disease severity, assessed by the Psoriasis Area and Severity Index (PASI, ranging from 0 to 72), which takes in account appearance and extension of the lesions. Most psoriasis patients present with a mild form of disease which is usually treated with topical agents, with local anti-inflammatory and/or anti-proliferative action. However, moderate and severe disease cases require systemic treatment. Systemic treatment usually comes after unsuccessful topical strategies, in a two-tiered approach where systemic therapy is used as a second-line treatment of moderate to severe psoriasis. Traditional systemic therapies aim at general immunosuppression and include the use of cyclosporine and/or methotrexate (MTX). In the past decade, a better understanding of disease immunopathogenesis has been successfully translated into new drugs, known as biologics, targeting key inflammatory mediators and currently representing an effective third-line therapy in moderate-to-severe psoriasis patients, unresponsive to non-biologic systemic agents. As of mid-2014, there are five biologics approved for the treatment of psoriasis, targeting either T cells or cytokines such as TNF or IL-12/IL-23 (see also Chap. 43) [155] (Table 21.2).
Table 21.2
Targeted therapy for Psoriasis in 2014
Mechanism of action | Name | Molecular target | Phase | Formulation | Administration route | Company | Reference |
|---|---|---|---|---|---|---|---|
Anti-Tcells | Alefacept | CD2 | Approved 2003 (US) | Human LFA-3/IgG1 fusion protein | IM or IV | Biogen | |
Anti-cytokine | Etanercept | TNF | Approved 2004 (US and EU) | Human TNF-R(p75)-lgG1 fusion protein | SC | Amgen | |
Infliximab | TNF | Approved 2006 (US and EU) | Mouse-human IgG1 chimeric monoclonal antibody | IV | Janssen Biotech | ||
Adalimumab | TNF | Approved 2007 (EU) 2008 (US) | Human IgG1 monoclonal antibody | SC | Abbott | ||
Ustekinumab | IL12p40 (IL-12,IL-23) | Approved 2009 (US and EU) | Human IgG1 monoclonal antibody | SC | Janssen Biotech |
Anti T-Cell and Anti-cytokines Biologic Drugs
The first biologics to be approved for psoriasis treatment were anti-T cell therapies targeting T cell adhesion or activation.
The first biologic to be approved was alefacept in 2003, a LFA-3/IgG1 fusion protein binding CD2 on T cells and thus, selectively inducing apoptosis of CD2(+) human memory-effector T cells [156]. Clinical efficacy was shown in phase III studies with 40 % of patients achieving a PASI75 response (75 % reduction of the PASI) [157] and more that 50 % achieving PASI 50 (50 % reduction of the PASI) [158]. A further antibody, anti-T cell strategy was approved in 2003 and consisted in a humanized antibody (efalizumab) binding and blocking CD11a, a key molecule for T-cell activation and migration through the circulation into the skin [159]. Despite a good efficacy profile, [160–166], efalizumab has been withdrawn from the market in 2009, due to three cases of progressive multifocal leukoencephalopathy [167], highlighting the importance of carefully monitoring the long-term safety of immunomodulatory therapies.
An alternative strategy to anti-T cell targeting aims at interfering with the psoriasis cytokine network using anti-cytokine biologic drugs. TNF blockade was serendipitously found to ameliorate psoriasis in a patient with IBD treated with the anti-TNF drug infliximab and had concomitant psoriasis [168]. Currently there are three anti-TNF biologics approved for psoriasis: etanercept, a human p75 TNF receptor fusion protein (approved in 2004), infliximab, a humanized chimeric anti-TNF monoclonal antibody (approved in 2006), and adalimumab, a fully human monoclonal antibody (approved in 2008). The efficacy of TNF inhibitors has been tested in phase III clinical trials, showing up to 80 % of treated patients achieving PASI75 within 10–12 weeks of treatment [169–176]. TNF neutralization causes early down- modulation of myeloid cell-related genes, with decrease of Th17 cell products and downstream molecules in just 2 weeks after commencing therapy [125, 177, 178]. The latest biologic to be approved for psoriasis in 2009, ustekinumab, is a monoclonal antibody simultaneously blocking the heterodimeric proteins IL-12 and IL- 23 via its binding to the shared subunit p40. Its efficacy is quite high, with 67 % of patients achieving PASI75 at 12 weeks of treatment [179, 180].
Some serious adverse events, such as opportunistic infections and reactivation of latent tuberculosis, have been reported for these monoclonal antibodies [181]. Long term safety data (up to 4 and 5 year treatment) are now available for etanercept and ustekinumab [169–171, 182, 183], suggesting a safe use of these drugs.
Emerging New Drugs: Biologics and Small Molecules in Clinical Trials
A number of other biologic drugs is currently being investigated in clinical trials, as of mid-2014 (see also Chap. 43) (Table 21.3). In line with the prominent role of IL-23/IL-17 axis in psoriasis, there are three antibodies (BI655066, tildrakizumab, and guselkumab) specifically targeting the IL-23p19 subunit which are currently being tested in phase II [184], III [185] or have recently completed phase II [186] clinical trials, respectively. Data from a small phase 1 study showed between 50 (10 mg) and 100 % (300 mg) of guselkumab-treated patients achieving PASI75, with improvements generally maintained through week 24 and rate of adverse event similar to placebo-treated patients [187]. Preliminary phase IIb data presented at the American Academy of Dermatology (AAD) meeting 2014 showed up to 81 % of patients receiving the highest dose achieving PASI75 response [188]. Phase 3 data for tildrakizumab presented at AAD 2013 showed PASI75 response rates of 64–74 % [189].
Table 21.3
Emerging drugs in clinical trials as of 2014
Type | Mechanism of action | Name | Molecular target | Phase | Formulation | Administration route | Company | Reference |
|---|---|---|---|---|---|---|---|---|
Biologics | Anti-cytokine | Tildrakizumab (MK-3222) | IL-23p19 | Phase III | Humanized IgG1 monoclonal antibody | SC | Merck | |
Guselkumab (CNTO 1959) | IL-23p19 | Phase II | Human IgG1 monoclonal antibody | SC | Janssen Biotech | |||
BI655066 | IL-23p19 | Phase II | Humanized IgG1 monoclonal antibody | SC | Boehringer Ingelheim | [184] | ||
Brodalumab (AMG 827) | IL-17R | Phase III | Human IgG2 monoclonal antibody | SC | Amgen | [193] | ||
Ixekizumab (LY2439821) | IL-17 | Phase III | Humanized IgG4 monoclonal antibody | SC | Eli Lilly | [190] | ||
Secukinumab (AIN457) | IL-17 | Phase III | Human IgG1 monoclonal antibody | SC or IV | Novartis | |||
PDE4 inhibitor | Apremilast (CC-10004) | PDE4 | Phase III | N/A | Oral | Celgene | ||
Small molecule | JAK inhibitor | Tofacitinib (CP-690,550) | JAK1 and JAK3 | Phase II | N/A | Oral | Pfizer | |
Tofacitinib (CP-690,550) | JAK1 and JAK3 | Phase II | N/A | Topical | Pfizer | [198] |
Monoclonal antibodies blocking either IL-17A (ixekizumab and secukinumab) [190–192] or IL-17R brodalumab [193], have shown remarkable efficacy in phase 2 clinical trials with more than 70 % of patients achieving PASI 75, and more than half achieving a striking PASI 90. While phase 3 clinical trials are currently ongoing, initial molecular data showed that the effect of IL-17 blockade on expression of genes synergistically regulated by IL-17 and TNF is greater than in previous studies with anti-TNF therapy [194].
Notwithstanding the efficacy of the biologic drugs currently available in the clinic, at least one third of patients do not respond to biologic therapy [195] or lose initial responsiveness, due to the development of anti-drug antibodies (ADA), which causes decreased drug efficacy and/or induction of adverse events [196, 197]. Moreover, biologic drugs pose a considerable economic burden due to their cost of about £10 k per patient/per year. Finally, a sizable number of patients with mild-to-moderate psoriasis still rely only on traditional topical treatments to manage their disease. Thus, other therapeutic options are being explored, such as small molecules, i.e. low molecular weight organic compounds targeting key molecules involved in cellular signalling. Based on the importance of cytokine- driven inflammatory pathways in psoriasis, the most promising small molecules currently under testing are targeting key cellular components in cytokine signalling, such as Janus kinases (JAK), as well as enzymes involved in cytokine production. Among JAK inhibitors, tofacitinib, which specifically inhibits JAK1 and 3 and is approved for RA treatment, has showed good efficacy in phase II trials for both its oral and topical formulation [198, 199] with 66.7 % of patients treated with the highest dose of oral tofacitinib reaching PASI 75 at 12 weeks in a phase 2b study [200]. Apremilast, a phosphodiesterase four inhibitor which inhibits an enzyme involved in the breakdown of cAMP, thus suppressing the production of pro-inflammatory cytokines is also been tested in Phase IIb clinical trials [201, 202] showing 41 % of patients achieving PASI75 at week 16 with the highest dose. A cheaper manufacturing process, a route of administration (oral or topical vs injectable) which ensures high patient compliance and an overall good safety profile, makes the use of these small molecules in mild-to-moderate psoriasis foreseeable
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


