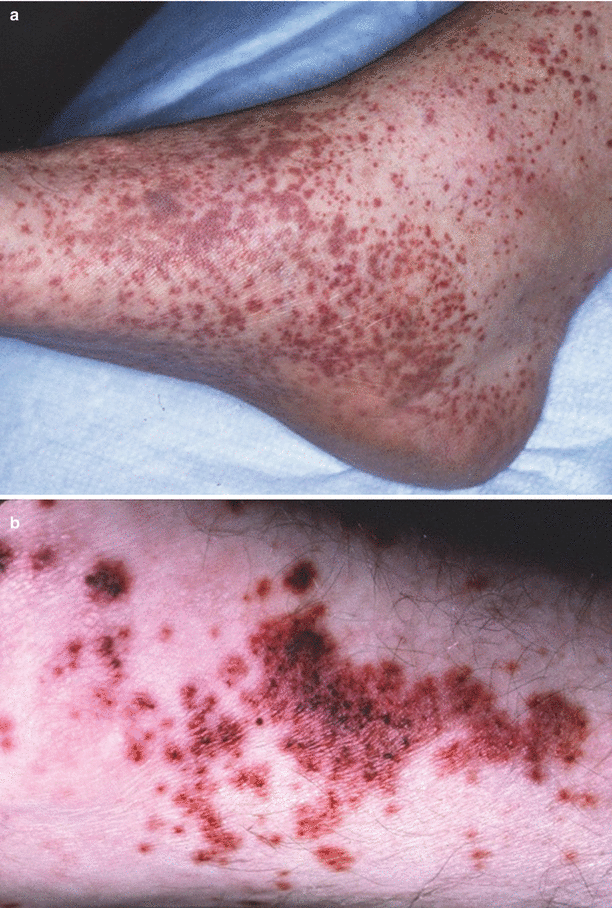
Fig. 34.1
Dermatitis herpetiformis. (a) Excoriated papules and papulo-vesicles on elbows and knees of a DH patient. (b) Urticarial plaques of DH
Patients with mild disease usually have involvement of the knees, elbows and dorsal forearms. Those patients with severe disease usually present with additional involvement of the trunk and extremities. Lesions also frequently occur in areas of pressure such as the belt line. Although the skin generally heals without scarring after resolution of the symptoms, post-inflammatory hyperpigmentation may occur [10].
Other uncommon cutaneous findings are petechiae on the fingers or palms, mainly occurring in children, but also found in adults [15, 16]. Occasionally, these palmar lesions are the primary manifestation of the disease [17].
DH may also occur in the oral mucosa; however, this has not been confirmed through direct immunofluorescence microscopy, and in some cases may have been aphthous stomatitis that is known to be an oral manifestation of CD [10]. Mucosal and tongue involvement may occur as vesicles, erosions or erythematous macules with or without discomfort. An additional association with celiac disease reveals itself in tooth enamel defects, such as horizontal grooves, pits or discoloration [18]. One study supporting this reported that in 30 adults with DH, 53 % had enamel defects compared with 2 % of 66 healthy controls [19]. One small study of ten children compared to healthy children controls showed eight had enamel defects while only 13 % of healthy children had similar defects [20].
Associated Diseases
The most common associated condition is autoimmune thyroid disease, with hypothyroidism being more likely [21–23]. One retrospective study of 264 adults with DH revealed that 11 % had thyroid disease [22].
Type 1 diabetes mellitus is also associated with DH with estimates between 2 and 5 % [10]. Pernicious anemia has a slightly higher frequency in patients with DH, ranging between 1 and 3 % [21].
An increased risk for non-Hodgkin lymphoma has reportedly been associated with celiac disease and this may also occur in DH [24]. One Swedish population-based study with 1354 patients with DH revealed a slight increased overall risk for malignancy (standard incidence ratio [SIR] 1.2, 95 % CI 1.0–1.4), due to increased cases of lymphoma and leukemia [25]. The effect of adherence to a gluten-free diet on the reduced risk for lymphoma was reported in one retrospective study in the United Kingdom; however, more studies are necessary to demonstrate the link between gluten-free diets and reduced risk of malignancy in DH. There is an increased risk of malignancy in CD, so it is likely that with a big enough sample size, a similar risk would be documented in CD’s cutaneous manifestation, DH [25].
Diagnosis
Confirming the diagnosis of DH through laboratory studies is essential, through histopathology of lesional skin, direct immunofluorescence microscopy (DIF) of perilesional skin and serologic studies. DIF is the gold standard test for diagnosis.
A 4 mm punch biopsy of a small intact vesicle is best for hematoxylin and eosin (H&E) staining. If there are no intact vesicles, involved erythematous skin is better than excoriated lesions, as these lesions may yield nonspecific results.
Findings on H&E staining vary with the age of the sampled lesion. Early lesions may show only a neutrophilic infiltrate, with or without eosinophils, and papillary microabscesses, which are neutrophils in the tips of the dermal papillae (Fig. 34.2a) [27]. After 48 h, lesions on H&E show subepidermal vesiculation at the dermal tips which over time bridge to form larger subepidermal vesicles, containing neutrophils, eosinophils and fibrin. Additionally, the dermis shows a perivascular lymphocytic infiltrate with neutrophils and eosinophils [27].
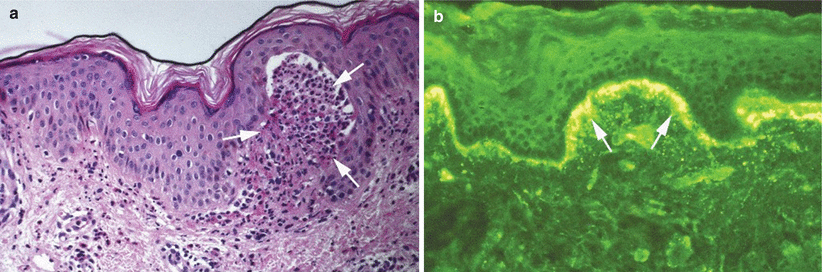
Fig. 34.2
Dermatitis herpetiformis. (a) Neutrophils in dermal papillary tips (arrows) on hematoxylin and eosin staining of biopsy from involved skin. (b) Granular IgA in the papillary dermis (arrows) with focusing of grains in dermal papillae from perilesional, clinically normal-appearing skin
The characteristics of DH on H&E histopathology are similar to other subepidermal bullous disorders. DH can be particularly difficult to distinguish from linear IgA bullous dermatosis, bullous systemic lupus erythematosus, and even bullous pemphigoid; however, numerous eosinophils are usually present in pemphigoid [27].
A punch biopsy for DIF must come from perilesional, clinically normal-appearing skin immediately adjacent to a lesion as biopsies taken from lesional skin are more likely to yield false negative findings [28]. The characteristic finding is granular deposits of IgA in the dermal papillae (Fig. 34.2b). DH may also show deposits of IgM, fibrinogen and C3 in a similar pattern [27]. A fibrillar pattern of IgA deposition is found occasionally rather than a granular one [29]. A retrospective study of 264 patients with DH found the DIF was positive in 92 % of patients (244) [21]. Of note, the DIF may be negative in patients on a strict gluten-free diet as this reduces IgA deposits in the skin. One retrospective series found that 10 of 41 patients with DH on a strict gluten-free diet after 13 years did not have detectable IgA deposition in the skin [30]. Treatment of DH with dapsone alone does not alter the findings of IgA deposition on DIF [10].
At times, granular deposits of IgA along the basement membrane appear on DIF, which may confound the diagnosis with linear IgA bullous dermatosis; however, the sharp linear IgA deposition usually separates the two disorders [6, 31]. In these instances, indirect immunofluorescence testing for basement membrane antibodies of linear IgA disease and serologic testing for IgA antibodies to TG2 and TG3 in DH may aid in clarification of the diagnosis. The same testing is useful in the extremely rare occasions when the DIF findings are equivocal or negative [32]. Additionally, these levels fall when a strict gluten-free diet is followed serving as a useful tool for monitoring response and adherence to this diet. Lastly, another useful serology in the evaluation of patients with DH is a serum total IgA level for detecting partial IgA deficiency, which occurs at an increased frequency in celiac disease. Such patients may not show IgA autoantibodies; therefore, IgG tissue transglutaminase and endomysium are better markers for this occasion [10].
Treatment
A strict gluten-free diet is the most effective therapy for patients with DH; however, remission can take months to years [33]. The most effective pharmacologic agent is dapsone, which can resolve skin lesions and pruritus within 48–72 h [33].
Dapsone therapy in adults is started at 25–50 mg daily and slowly increased to 2 mg/kg daily based on response and tolerance to therapy, in association with adherence to a gluten-free diet. Adults on unrestricted diets may need between 50 and 150 mg daily for complete response [34]. Even at optimal dosing, mild eruptions of one to two new lesions per week are possible. These events do not warrant an increase in dose, rather a potent topical corticosteroid may be applied to the new lesions.
As dapsone is a sulfone antibiotic, mild asymptomatic side effects of this medication are expected in nearly all patients. Dapsone competitively inhibits dihydropteroate synthase, an enzyme involved in folic acid reduction; therefore, most patients will have some red blood cell (RBC) hemolysis. Patients with glucose-6-phosphate deficiency (G6PD) should not take dapsone, as they are at increased risk for severe hemolytic anemia from oxidative stress. Testing for this deficiency is recommended before starting therapy.
Methemoglobinemia is another dose-related side effect of dapsone therapy. Methemoglobin, an oxidized form of hemoglobin, has a reduced ability to carry oxygen, leading to symptoms of hypoxia and anemia. Cardiovascularly-compromised patients may have symptoms even at low doses. Supplementation of these patients with antioxidants such as Vitamin E may theoretically help mitigate these effects.
Additional potential side effects include agranulocytosis and a hypersensitivity reaction. Agranulocytosis generally appears 2–12 weeks after commencement of dapsone. Surveillance through lab monitoring will detect early signs of this potentially lethal side effect, allowing discontinuation of the drug. Dapsone-associated hypersensitivity symptoms are a morbilliform cutaneous eruption with flu-like symptoms, including fever, lymphadenopathy, hepatitis and eosinophilia.
Because dapsone is associated with potential serious side effects, careful laboratory monitoring is crucial. Initial lab screening should include a complete blood count (CBC), liver and renal function panels, and G6PD screening. After initiation of dapsone, interval lab screening is recommended: CBC every 1–2 weeks for 1 month then every 1–3 months for 6 months; thereafter CBC and liver function should be checked every 3–6 months if dapsone is not increased during this time frame.
After 2–3 months of dapsone therapy and a gluten-free diet, dapsone can be tapered slowly. Reduction of 12.5 mg every 2–4 weeks is recommended.
For patients who are intolerant of dapsone, there are other sulfa-based drugs available, sulfasalazine and sulfapyridine. Sulfapyridine is not available commercially in the United States, but can be obtained through compounding pharmacies. Sulfasalazine is metabolized to sulfapyridine in the intestine; however, the level of active metabolite is more predictable when sulfapyridine itself is given [35]. Sulfapyridine dosing should be started at 0.5 g three times per day and increased up to 6 g daily for control of symptoms. Sulfasalazine should be dosed between 1 and 2 g daily [35]. Both medications have the potential adverse effects of agranulocytosis and hypersensitivity reactions, but not hemolysis. Adequate fluid intake and possible alkalinization of the urine with oral bicarbonate is also recommended to reduce the risk of drug-induced nephrolithiasis. As with dapsone, lab monitoring (CBC, LFTs and urinalysis) is recommended periodically.
Although adherence to a strict gluten-free diet is challenging, as gluten is hidden in many common foods, patients will benefit from this and will be able to decrease or discontinue medications. A 36-month study of 81 patients with DH on a gluten-free diet for 6–36 months and 49 patients with DH on a normal diet reported that 93 % of patients on a gluten-free diet achieved reductions in dapsone dosing versus 16 % of patients on a normal diet [36]. There are many resources for patients about gluten-free diets, including the Celiac Disease Foundation (www.celiac.org) and the Gluten Intolerance Group (www.gluten.net).
An additional adjunctive treatment for the pruritic skin lesions of DH is potent topical corticosteroids. These topicals are not effective monotherapy, and systemic glucocorticoids are generally ineffective.
Treatment of Children
Management of children is the same as for adults. They should follow a gluten-free diet and if necessary take dapsone 0.5–2 mg/kg/day [35]. As with adults, dapsone may be tapered as the gluten-free diet controls the disease.
Prognosis
Dermatitis herpetiformis is a chronic condition that requires life-long adherence to a gluten-free diet or treatment with dapsone. When gluten is re-introduced into the diet, symptoms may recur within weeks to months and when dapsone is discontinued, symptoms may appear within 2 days [37]. A small percentage of patients (10–15 %) may maintain remission despite the discontinuation of both dietary and pharmacological therapy [38].
Linear Immunoglobulin A Bullous Dermatosis
Linear IgA bullous dermatosis (LABD) or linear IgA disease is a rare, acquired idiopathic or drug-induced autoimmune subepidermal blistering disease. The main feature of this disease is the linear deposition of IgA at the dermo-epidermal junction. Distinguishing this from DH can be difficult, since similar clinical and histopathologic findings may occur; however, LABD is rarely associated with gluten-sensitive enteropathy and its sharp linear IgA deposition is easily distinguished from the granular IgA of DH by the experienced immunopathologist [39].
LABD occurs in both adults and children. In children, the disorder once known as chronic bullous disease of childhood is now recognized as the childhood form of LABD [40]. Adults may present with tense vesicles and bullae within erythematous annular plaques (Fig. 34.1b) or like DH as excoriated papules on the extremities, buttocks and face. Different from adults, children present clinically with widespread annular lesions with peripheral vesiculation on the lower abdomen, thighs and groin area [41]. The oral mucosa can be involved in both adults and children.
Epidemiology
Reported incidence rates range from less than 0.5–2.3 cases per million individuals yearly [42]. No predilection based on ethnicity or gender for LABD has been established [42]. LABD rarely occurs in neonates [43]. It can develop in children between the ages of 6 months and 10 years. The average age of onset in 25 affected children was 4.5 years [40]. Adults present with LABD later in life, with many cases occurring after age 60 [40, 42]. Drug-induced cases may occur at any age.
Risk Factors
There is no known inciting factor for most cases of LABD. Medications are suspected in multiple cases. The most common drug implicated is vancomycin [42]. Other reported associations are listed in Table 34.1.
Table 34.1
Medications implicated in precipitating linear IgA bullous dermatosis
Drugs associated with linear IgA bullous dermatosis | |
|---|---|
Common | Vancomycin |
Multiple reports | Captopril |
B-lactams | |
Cephalosporins | |
NSAIDs | |
Phenytoin | |
Isolated reports | Acetaminophen |
Amiodarone | |
Atorvastatin | |
Benazepril | |
Candesartan/eprosartan | |
Carbamazepine | |
Furosemide | |
Gemcitabine | |
Interleukin-2 | |
Lithium | |
Somatostatin | |
Trimethoprim/sulfamethoxazole | |
Pathogenesis
A search for target autoantigens in LABD has been complicated, as studies of patient sera have shown varied results. LABD can be initially divided into subtypes based on ultrastructural location of IgA (sublamina densa type and lamina lucida type); however, overlap between the two types may occur. These can be established using basement membrane zone human salt-split skin and indirect immunofluorescence in cases where there is circulating IgA basement membrane antibody, since such separation occurs in the lower lamina lucida. Lamina lucida antigens will adhere to the epidermal side of the basement membrane separation and sub-lamina densa reactive antibodies will bind to the dermal side. In cases without circulating IgA basement membrane antibody, immunoelectron microscopy or basement membrane separation of the DIF-positive biopsy can be used to identify the type. In most cases this is unnecessary since treatment for both variants of LABD is the same.
The lamina lucida type predominantly targets a 97-kDa antigen and a 120-kDa antigen that are the proteolytic fragments of the extracellular portion of the bullous pemphigoid antigen 2 (BP180), a key epidermal-dermal adhesion transmembrane protein [44, 45]. Less frequently, LABD is associated with the NC16a epitope of BP180 [46–48]. The sub-lamina densa type of LABD has been reported to be predominantly type VII collagen, although a number of other antigens have been proposed [49]. Lastly, there is a subset of patients with features consistent with LABD who have both IgA and IgG antibodies against the basement membrane zone. A Japanese review of 213 patients with LABD found both antibodies in approximately 20 % of the cases [50].
Both humoral and cellular immunity may contribute to the pathogenesis of the lesions. Skin and mucosal findings may be the result of an antibody-induced local inflammatory response from the release of proteolytic enzymes by neutrophils and other inflammatory cells [42].
Clinical Manifestations
Patients generally present with lesions on the skin, or on the mucous membranes, or on both locations. Blister formation is sub-epidermal; therefore, the vesicles and bullae are typically tense, rather than flaccid as is found in pemphigus. Similar to DH, pruritus and scratching may leave only excoriations and erosions.
Adults and Children Manifest the Disease Differently (Fig. 34.3a–d)
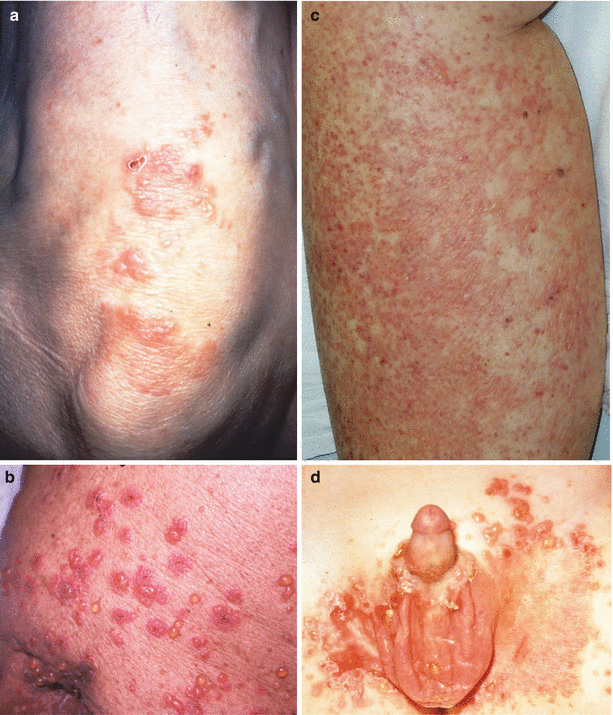
Fig. 34.3
Linear IgA bullous dermatosis. (a) Papulo-vesicles on the extensor forearms resembling DH. (b) Scattered tense vesicles with associated urticarial lesions. (c) Diffuse erythema and superficial vesicles in a case of vancomycin-induced LABD. (d) Scrotal and inguinal vesicles in childhood LABD
Children often present with acute development of vesicles or bullae on sites of erythematous or normal skin. The distribution of these lesions is generally widespread, involving the face (usually the perioral area), trunk, genitalia, hands and feet. The lower abdomen, perineum and inner thighs may be the most intensely involved areas [40, 51, 52]. At the periphery of resolving lesions, new blisters often form, resulting in an arciform or annular configuration. These lesions are classically known as “string of pearls”, “crown of jewels” or “rosettes” [53]. Children usually have pruritus, which can be severe, while other affected children may be asymptomatic. For some children, intense pruritus heralds relapse of their condition [54, 55].
Adults with LABD present typically with acute onset of skin lesions, rather than a gradual onset [41]. The lesions arise on uninvolved skin or within erythematous plaques. Adults generally do not develop the string of pearls lesions with peripheral vesiculation, but this may occur [41]. The general distribution of lesions in adults involves the face (also the perioral area, like children), trunk, extensor extremities and buttocks [42]. With the distribution including the extensor extremities, this disease can be difficult to distinguish from DH. Additionally, localized variants of LABD presenting as limited eruptions of bullae or annular erythematous plaques have been reported in several different case reports [20, 56–62]. As with children, adults can also experience intense pruritus resulting in the development of excoriated papules or prurigo nodularis-like lesions [63, 64].
Mucosal Involvement
Adults and children can develop mucous membrane involvement. One study reported that up to 80 % of adults have mucosal lesions [40]; however, in children, estimates of mucosal involvement are varied. One study with 25 children from the United Kingdom reported 64 % had mucosal lesions [65]. Alternatively, two retrospective studies of similar sample size from Tunisia and Japan reported only 8–3 % involvement, respectively [66].
Infrequently the mucosa is the sole manifestation of LABD [67–70]. In these cases, mucosal-predominant LABD is considered a form of cicatricial mucous membrane pemphigoid [48].
Erosions or ulcers are the primary mucosal lesions as it is rare to find intact vesicles or bullae. Affected sites include any mucosal surface of the body, including the ocular conjunctivae, oro- and nasopharynx, larynx, esophagus, vagina and anus, with the ocular and oral mucosae being the most common sites [40–43]. Within the oral mucosa, the lesions are frequently found on the palate, palatine arches and buccal mucosae [42]. Additionally, erosive gingivitis and cheilitis may occur as manifestations of mucosal LABD [40, 70]. Ocular symptoms manifest as erythematous conjunctivae, discharge, pain or a foreign body-like sensation [65]. Occasionally, patients develop symblepharon and ectropion, thus the disease is essentially an IgA variant of ocular pemphigoid [71]. Mucosal scarring can lead to serious adverse sequelae, including corneal damage leading to blindness, airway obstruction and esophageal strictures [71–73].
Drug-induced and Idiopathic LABD
Medications are often causes of LABD. Vancomycin is the most common offending medication; however, over 20 other medications have also been linked to it. Other antibiotic classes in this list are beta-lactams and cephalosporins. Nonsteroidal anti-inflammatory medications and acetylcholinesterase inhibitors, such as captopril, have been cited in case reports (Table 34.1). The idiopathic form of LABD does not differ significantly from the drug-induced form [42]. Presentations of both of these forms of LABD can be with localized involvement rather than widespread. They may or may not involve the mucosa [42]. They can be morbilliform eruptions. They can even resemble erythema multiforme or toxic epidermal necrolysis [29, 74–79]. Therefore, one should consider drug-induced LABD in the differential diagnosis when evaluating a patient with possible toxic epidermal necrolysis.
The similarities between drug-induced LABD and idiopathic LABD were evaluated in a retrospective study of 16 patients presumed to have spontaneous LABD and 12 patients presumed to have drug-induced LABD. This study found closely associated frequencies in both groups of erythematous plaques, string of pearls-like configurations, target lesions and mucosal involvement [29]. The drug-induced group contained a higher frequency of patients with atypical presentations of large erosions and positive Nikolsky signs.
Adults are more frequently afflicted with drug-induced LABD; however, it has also been reported in children [20, 40, 42]. The onset of lesions generally begins within the first month of drug administration and then they resolve gradually over the ensuing several weeks [20, 80, 81]. Some patients may have the lesions persist beyond this timeframe. Furthermore, if the patient is re-exposed to the inciting medication, rapid reappearance of the lesions can occur [82].
Associated Disorders
The most common non-malignant disorder associated with LABD is ulcerative colitis (UC) [50, 83–87]. Two retrospective studies reported this possible association, one from the United Kingdom and one from Japan. The study from the UK found that of 70 patients, 5 patients (7 %) had LABD, and UC preceded the diagnosis of LABD by an average of 6 years [85]. The review from Japan of 213 cases of LABD found four patients with UC [50]. This association is not well elucidated. Some authors suggest that abnormal IgA1 production by the inflamed bowel may contribute to the development of LABD; however, even after removal of the colon, some patients continue to have recurrences of LABD, while others have the disease completely resolve [83, 85, 87].
Malignant disorders, lymphoproliferative and solid organ types have also been associated with LABD in multiple case reports [31, 58, 88–104]. Despite these reports, no retrospective analyses have been performed to confirm this association. Further studies may confirm this suspected link between LABD and certain malignancies.
Diagnosis
The histopathologic findings with hematoxylin and eosin staining of involved skin are identical to those of dermatitis herpetiformis, showing lymphocytes, eosinophils and papillary microabscesses [42, 55] (Fig. 34.4a). Other characteristic findings are a subepidermal blister with a diffuse underlying neutrophilic infiltrate in the dermis.
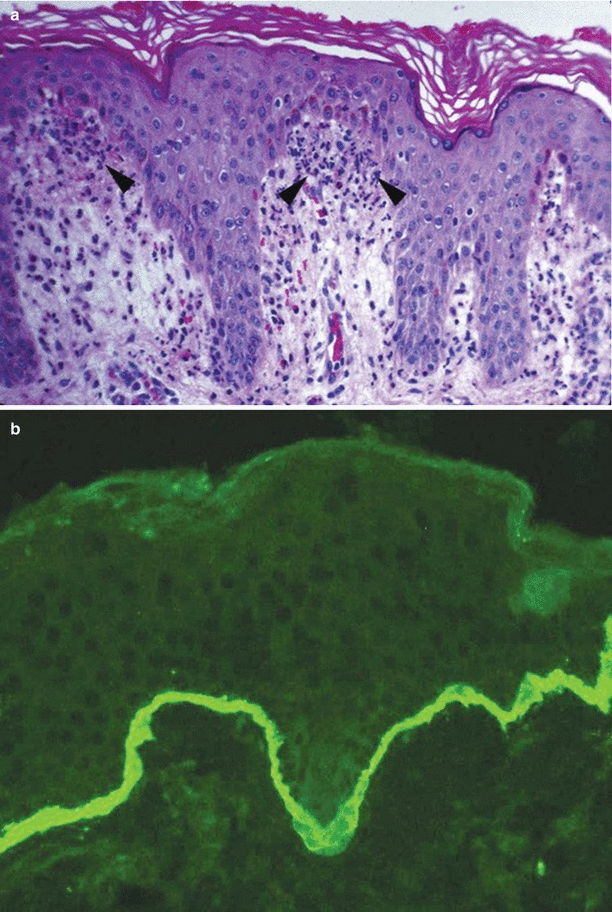
Fig. 34.4
Linear IgA bullous dermatoses. (a) Dermal papillary edema with a neutrophilic infiltrate (arrowheads) similar to DH. (b) Characteristic sharp linear IgA antibody deposition along the basement membrane zone
A biopsy of perilesional skin demonstrating linear deposits of IgA along the basement membrane zone on direct immunofluorescence (DIF) testing is the gold standard test for diagnosis (Fig. 34.4b) [55]. Indirect immunofluorescence may be positive. Antibody binding on indirect immunofluorescence with basement membrane zone human salt-split substrate to the epidermal side of the induced cleavage zone is the most common location, while binding to the dermal side may occur in older patients or those who have both IgA and IgG deposits on the epidermal side of the basement membrane zone [50].
Treatment
The first-line medication therapy for LABD, as with DH, is dapsone [42, 52, 54, 55, 109]. Dapsone is started at a low dose, less than 0.5 mg/kg daily for children and 25–50 mg in adults. Gradually this dose is titrated upward over several weeks, depending on tolerance and treatment response [110]. This response can be immediate, resolving lesions within a few days of initiating therapy; however, some patients with more extensive disease and incomplete response to dapsone may need oral corticosteroids to accelerate improvement and effectively suppress the lesions [41, 54, 66, 111]. When patients need longer periods of immunosuppressive therapy or patients with predominantly mucosal involvement need more aggressive therapy, steroid-sparing agents can be effective, such as mycophenolate mofetil, azathioprine, intravenous immunoglobulin (IVIG), cyclophosphamide and rituximab [57, 111–122].
For patients who cannot tolerate dapsone, sulfapyridine or sulfamethoxypryridazine may be effective second-line therapies. Evidence for this is limited to reports from specialists in the field, as prospective therapeutic trials have not been performed to confirm this [42, 54, 55, 110, 123]. Sulfamethoxypyridazine is not available in the United States and sulfapyridine is only available through compounding pharmacies. The adult dosing range is 1000–1500 mg daily of either agent [54]. The sulfapyridine dosing range for children is 15–60 mg/kg daily [42, 55]. There is not an established dosing range for sulfamethoxypyridazine in children [42]. Both of these medications have been used in combination therapy with dapsone [55, 123, 124].
Colchicine can be effective in children with LABD. Some case reports and case series have shown it to be a reasonable substitute therapy for dapsone [111, 125–127]. In a series of eight children with systemic glucocorticoid-refractory LABD, the addition of colchicine led to dramatic improvement in five patients within 4–6 weeks. Furthermore, these children were able to taper off steroid therapy. The typical dosage in children is 0.6 mg twice daily.
Adults have also responded to colchicine according to some reports [56, 128]; however, other authors have not seen such reported results [54]. The adult dose range for colchicine is 0.6–1 mg two to three times daily [42, 52, 111, 127].
Additional therapies that have been used for LABD include tetracycline in combination with nicotinamide and topical tacrolimus ointment. This therapeutic combination has been effective for the treatment of bullous pemphigoid and has been applied to LABD. Three adult patients with LABD reported disease resolution within a few weeks of starting therapy [129–131]. The dosing range for tetracycline and nicotinamide are 1000–1500 mg daily and 900–2000 mg daily, respectively. Children under age nine cannot take tetracycline due to the adverse effect on developing teeth. Additional studies will help to confirm the efficacy of this treatment.
Children with LABD may respond to systemic antibiotic therapy. The mechanism for efficacy is not known, whether it is due to anti-inflammatory or antibacterial properties, or another unknown action remains to be clarified. One case series reports that in seven children treated with flucloxacillin, all had complete resolution, but only four children stayed in remission off of therapy [132]. Additional antibiotics reported to effectively treat LABD in children are oxacillin, dicloxacillin, erythromycin, micocamycin and trimethoprim-sulfamethoxazole [48, 66, 133–139].
Potent topical corticosteroids may be used adjunctively to accelerate resolution of lesions on the trunk or extremities, while low potency topical steroid creams can be used on the face, genitals or intertriginous areas.
Lastly, drug-induced LABD typically resolves when the inciting agent is stopped; however, in severe or persistent cases, dapsone and/or prednisone may be used to achieve faster resolution. Therapy should be tapered off early in the treatment course, within 4–6 weeks, to ascertain whether the disease is still active, warranting continuation of systemic therapy. A prolonged treatment course rarely occurs in this disease.
Prognosis
Idiopathic LABD can persist from months to several years in adults, whereas in children, it typically resolves before puberty [40, 52, 54]. This disease can also prevail for a decade or even longer. It can also recur after long periods of remission [54]. This is in contrast to drug-induced LABD, which usually improves within a few days of cessation of offending drug and resolves within several weeks [80].
The treatment duration for idiopathic LABD is variable. Therapy is generally continued for several weeks after complete resolution of lesions and then gradually tapered off. If at any point lesions recur, the treatment medication should be restarted [54].
Cutaneous lesions typically heal without scarring; however, mucosal lesions may lead to stricture formation or conjunctival and corneal scarring. These sequelae can have a significant impact on patients’ oral hygiene and nutritional status.
Immunoglobulin A Pemphigus
IgA pemphigus, with subtypes subcorneal pustular dermatosis-type IgA pemphigus and intraepidermal neutrophilic IgA dermatosis, is a blistering disorder characterized by autoantibodies against the desmosomal components of keratinocytes. Subcorneal pustular dermatosis (SPD) appears clinically to resemble Sneddon-Wilkinson disease and IgA cell surface staining locates to the upper epidermis on DIF. The intraepidermal neutrophilic IgA dermatosis (IEN), as the name implies, exhibits IgA cell surface staining throughout the entire epidermis [140].
Epidemiology
Pathogenesis
In contrast to IgG-mediated pemphigus vulgaris, IgA pemphigus is an IgA-mediated anti-keratinocyte cell surface autoantibody disorder [142]. As stated above the target antigen in the SPD type is desmocollin 1, a calcium-dependent transmembrane glycoprotein of the cadherin family within the desmosomes [142, 143]. The target antigen in IEN has not been completely elucidated and studies have shown varying targets. Several patients have shown autoantibodies against desmoglein 1 and 3; however, when viewed through immunoelectron microscopy, the targets have been unidentified non-desmosomal transmembrane proteins [142, 144–147].
Clinical Features
Both types of IgA pemphigus are characterized by the subacute development of vesicles that evolve into pustules on erythematous plaques, usually distributed across the trunk and proximal extremities [140]. Other sites of involvement are the scalp, postauricular skin and intertriginous areas, while the mucous membranes are generally spared [48]. Pruritus may or may not be present, but can be pronounced if it is. Patterns of configurations may be herpetiform, annular or circinate [140]. Because the SPD type of IgA pemphigus so closely resembles Sneddon-Wilkinson disease, DIF must be performed to distinguish the two disorders.
Diagnosis
The diagnosis of IgA pemphigus is made through all tools available to the clinician, including clinical, histological, immunopathological and serological findings. Histological features are intraepidermal pustules and clefts with microabscesses in the subcorneal region for SPD and throughout the epidermis for IEN. There is a neutrophilic infiltrate in the epidermis and dermis with sparse or no acantholysis, especially in IEN [27].
DIF studies of perilesional skin reflect H&E findings in that intercellular IgA staining is located in the upper epidermal layers in SPD and throughout the epidermis in IEN. Weak intercellular IgG and/or C3 deposits may also be present [140]. IIF studies on monkey esophagus show expected intercellular IgA deposits, with testing positive about 50 % of the time [140].
Additional tests used to identify circulating IgA autoantibodies to desmocollin 1 are immunoblotting, ELISA using recombinant desmocollin, and immunofluorescence molecular assay using desmocollin-transfected COS-7 cells (monkey fibroblast-like kidney cells), which are performed only in research laboratories at this time [48, 143, 148]. In one series of 22 patients, ELISA tests showed positive autoantibodies to desmoglein 1 in three patients and desmoglein 3 in one patient [142]. All four patients had either the IEN subtype of IgA pemphigus or even clinical and histological features of pemphigus foliaceus. Ten of the 22 patients had the SPD subtype, and they did not have autoantibodies to the desmogleins, rather they had autoantibodies to desmocollin 1 in COS-7 cells [142].
Treatment
As with all other IgA dermatoses, dapsone is the first-line therapy in both subtypes of IgA pemphigus; however, it is not always effective and response to treatment is varied, with the SPD type being more resistant to therapy [149]. Dapsone doses range between 75–100 mg for initial disease control and may be tapered down for maintenance therapy. Colchicine may also be used if dapsone is not well tolerated [150]. The SPD subtype is generally more recalcitrant to treatment. In these cases, isotretinoin or acitretin may be used alone or with dapsone as combination therapy [151]. For patients with the IEN subtype who fail dapsone monotherapy, oral steroids may be added in concert with dapsone. Occasionally, patients may need more aggressive immunosuppressive therapy with a monoclonal antibody, adalimumab, or mycophenolate mofetil [140, 152]. High potency topical corticosteroids may be used adjunctively with oral therapy.
Immunoglobulin A Vasculitis (Henoch-Schönlein Purpura)
Henoch-Schönlein Purpura (HSP), now also defined as IgA vasculitis (IgAV), is a leukocytoclastic small vessel vasculitis of the dermis that occurs most commonly in children after a precipitating infection or drug reaction [153]. The hallmark manifestations of the disease are the tetrad of palpable purpura, arthralgias, abdominal pain and glomerulonephritis. Although IgA deposition can occur in other isolated cases of small vessel vasculitis and secondarily in other associated diseases with small and large vessel vasculitis involvement, such as systemic lupus erythematosus, Sjögren’s syndrome, rheumatoid arthritis and polyarteritis nodosa, HSP is the only disease that occurs because of the primary deposition of IgA complexes in the vessel walls [154–156]. Its manifestations are caused by this IgA deposition. The disease is generally self-limiting; however, protracted cases do occur and recurrences can happen. Adults can also manifest the disease after an inciting infection or drug reaction or for no identifiable reason.
Epidemiology
Children develop HSP most frequently between the ages of 3 and 12 years [157]. The annual incidence rates range from 3 to 26.7 per 100,000 for children and infants and from 0.8 to 1.8 per 100,000 for adults [157]. A population-based study from the United Kingdom showed a peak incidence for children between the ages of 4 and 7 years old. Adults appear to contract the illness between 45 and 50 years old; however, it has occurred in patients up to the age of 86 years [158–160].
HSP shows a slight predilection for males, with male-to-female ratios of 0.9–1.8 in children and 1.7–2.4 in adults [157–160]. Ethnic population studies reveal an increased frequency in white and Asian children and decreased frequency in blacks [157, 161]. Additionally, patients with familial Mediterranean fever have a significantly higher incidence rate for HSP [157, 162, 163].
Many epidemiological studies have shown a seasonality to the disease in children, with a prevalence for the fall and winter seasons and abatement in the summer [157]. This trend may be due to the association of HSP with increased winter-triggered upper respiratory infections in children [159]. Studies show that 30–65 % of IgA vasculitis cases develop after an upper respiratory infection [157, 164–168]. This seasonal association is not borne out in studies of adult cases; furthermore, no clear variations to the disease incidence have been illuminated [157]. Some epidemiologic studies have shown a possible association with preceding or concurrent malignancies; however, further studies are necessary to elucidate this possibility [157, 169–173].
Genetic factors have been investigated for predisposing and protective susceptibilities in the human leukocyte antigen (HLA) region. Two independent patient cohorts found an increased risk of HSP in patients with HLA-DRB1*01, HLA-DRB1*11, HLA-B35 and HLA-A11 alleles, while patients with HLA-DRB1*07 alleles in Spanish and Italian populations were found to be protected from HSP [6, 47, 157, 174–176].
Pathogenesis
IgAV is classified as a leukocytoclastic vasculitis due to the involvement of small vessels within the papillary dermis, generally the postcapillary venules. Neutrophils and monocytes are the main inflammatory infiltrate. Although the definitive role of IgA in the pathogenesis of HSP remains undefined, IgA deposition occurs within the small vessels of the affected organs, as evidenced on histological examination. Immunofluorescence staining reveals IgA, C3 and fibrin in these vessels. Additionally, infections and medications are the purported triggers for HSP; however, this has also not been completely clarified. Many studies have shown that immunologic, genetic and environmental components are involved [177–179].
IgA1, predominant in serum, is the main IgA antibody present in HSP. One theory regarding why this is found posits that abnormal glycosylation of the IgA1 hinge region would allow aggregation and large complexes to form [180]. Additional theories from research regarding the pathogenesis of IgAV include a role for IgA anticardiolipin antibodies, IgA rheumatoid factor antibodies and beta-2 glycoprotein 1 antibodies; however, more studies are needed to validate these potential contributors [181–185].
Clinical Features
The classic tetrad of HSP is palpable purpura, abdominal pain, arthralgias and glomerulonephritis. The palpable purpura generally occurs on the dependent areas of the body, starting on the bilateral lower extremities and extending up to the buttocks and back. Development of all the features is usually a subacute onset over days to weeks, with purpura and arthralgias presenting initially.
Skin manifestations are present in nearly 100 % of the presentation; however, the purpura may not always be the first sign. The rash generally appears with petechial, macular or even urticarial lesions that evolve shortly into palpable purpura, some with necrotic centers. Retiform plaques of purpura are common and characteristic (Fig. 34.5a, b). Sometimes, acral, scalp and facial edema accompany the rash.