Fig. 42.1
The SCC immune microenvironment. The SCC microenvironment involves a complex interplay of immunoinhibitory and immunostimulatory cells and cytokines. While Langerhans cells and plasmacytoid DCs are producing proinflammatory cytokines IL-15 and IFN-α respectively to enhance the immune response, the presence of tumor associated macrophages and regulatory T cells contribute to immune dysfunction and tumor invasion through the production of matrix metalloproteases (MMP9 and 11), pro-angiogenic factors (VEGF-C) and immunoinhibitory cytokines (IL-10, TGF-β, VEGF-A). These protumoral cytokines can also functionally compromise the other immune cells of the microenvironment, leading to immature myeloid dendritic cells, which express CD200, and Langerhans cells which express a mixed gene profile of immune activation and immune tolerance genes
The evidence of immunosuppression in SCC continues with the lack of particular immune cells in the local environment; NK cells, B cells and monocytes are rarely detected around SCC [8]. Each of these cells has been shown to play a role in tumor cell eradication. NK cells are lymphocytes that were first identified for their ability to kill tumor cells without deliberate immunization or activation [9]. Monocytes exhibit considerable selective cytocidal activity against tumor cells through the generation of reactive oxygen species (ROS) [10]. Though some studies have shown resting B cells to inhibit T-cell mediated regression of tumor cells, other studies have shown the key role that activated B cells play in T cell activation and creation of long term responses against cancer [11]. The collective scarcity of these cells in SCC may play a role in SCC‘s ability to establish itself, as well as grow and metastasize.
The presence of the myeloid-derived suppressor cells (MDSCs) provides further evidence and mechanisms for SCC- related immunosuppression. MDSCs are potent suppressors of T-cell mediated responses, partially due to the downregulation of E-selectin on vascular endothelial cells. The lack of E-selectin on endothelial cells restricts T-cell entry into tumors and is caused by the presence of nitric oxide (NO) in the microenvironment. SCC-infiltrating MDSCs have been shown to produce NO, thus contributing to the immune evasion of the tumor. A possible therapy of suppressing NO production through inhibiting inducible NO synthase (iNOS) has been shown to induce E-selectin expression in vitro and may be effective as a future therapy for SCC [12].
Tumor-Associated Macrophages in SCC
Macrophages are a major population of leukocytes that are found surrounding and infiltrating solid tumors [13]. In SCC, macrophages have been shown to be far more abundant than in normal skin [14]. These peritumoral and penetrating macrophages, as mentioned earlier, are defined as tumor-associated macrophages (TAMS) [15]. TAMS play a significant role in tumor behavior, with potential to inhibit or stimulate tumor growth. Some studies have shown TAMs are capable of eradicating tumor cells in vitro, yet others have correlated a poor prognosis with increased numbers of TAMS [16–18]. In their role in aiding and abetting the tumor, TAMS can fail to identify tumor antigens and can release angiogenic and tumor stimulating factors [19, 20]. It is suspected that the tumors themselves may be creating a dynamic microenvironment that transforms the TAMS into macrophages that allow for tumor growth [21].
Despite the potential ability of TAMS to inhibit tumor progression, an increased number of TAMS is correlated with a negative prognosis. This can be explained by a number of factors. In studies, TAMS have been shown to make matrix metalloproteinases (MMP)9 (gelatinase B) and 11 (stromelysin-3), zinc-dependent proteinases that participate in extra-cellular matrix degradation, which allow direct tumor invasion, as well as release pro-angiogenic factors otherwise sequestered within the extra-cellular matrix [14, 22]. This is in contrast to macrophages in normal skin, which produce these factors at lower levels than TAMS in the SCC microenvironment.
The macrophages of the SCC microenvironment have also been shown to be predominantly M2 macrophages, or alternatively activated macrophages. These macrophages are induced by IL-4, in contrast to M1 macrophages, or classically activated macrophages, which are induced by IFN-γ. A strong M1 macrophage response is thought to prevent tumor growth. In contrast, M2 macrophages have a lower antigen presenting capacity and have been positively correlated with tumor genesis and progression through inflammation. Recent studies have shown SCC- associated macrophages to be heterogeneously activated, with some expressing M1 markers, some expressing M2 markers and some expressing both M1 and M2 markers simultaneously [14]. This heterogeneous activation of TAMS in SCC gives rise to a potential therapy in driving TAM activation to the M1 anti-cancer phenotype.
TAMS have been shown to play a role in lymphangiogenesis, with the expression of vascular endothelial growth factor-C (VEGF-C) [23]. VEGF-C, a critical lymphangiogensis mediator, promotes increased lymph vessel density and has been correlated with increased risk of metastasis in squamous cell carcinomas of the oral cavity and melanoma [24, 25]. The increased lymph vessel density spurred by VEGF-C is an important factor in cancer’s development and spread.
With production of high levels of MMPs, expression of lymphangiogenic mediator VEGF-C and M2 characteristics, TAMS in SCC are failing to prevent tumor creation and progression and are rather promoting its survival and advancement. The overall behavior and subtype of TAMS in SCC show the immunosuppressive potential of immune cells when influenced by the tumor environment.
Dendritic Cells and SCC
Dendritic cells (DCs), the most potent antigen-presenting cell, exist in a variety of subtypes, and regulate the adaptive immune system [26, 27]. They are key players in cancer immune surveillance, with their ability to stimulate tumor-specific T-cell responses, along with having been shown to infiltrate various human tumors [28–31]. DCs are abundant in the skin immune system, existing mainly as epidermal Langerhans cells (LCs) and dermal myeloid DCs. LCs, with their epidermal localization, should be the first antigen presenting cell to encounter SCC tumor antigen, as SCC is a malignant proliferation of epidermal keratinocytes.
However, studies have shown that DCs from human cancers are often functionally compromised, with a decreased ability to induce IFN-γ and stimulate T-cells [32, 33]. Further studies have shown that the tumor microenvironment contains immunosuppressive cytokines which impair DC differentiation and function [26, 34].
With regards to DCs, SCC have been shown to have a relatively immunosuppressed microenvironment, with a lack of mature dendritic cells in comparison to normal skin (Fig. 42.2) [35, 36]. This is seen by the down-regulation of mature dendritic cell marker gene CD83 and decreased numbers of CD83 cells in SCC [6].
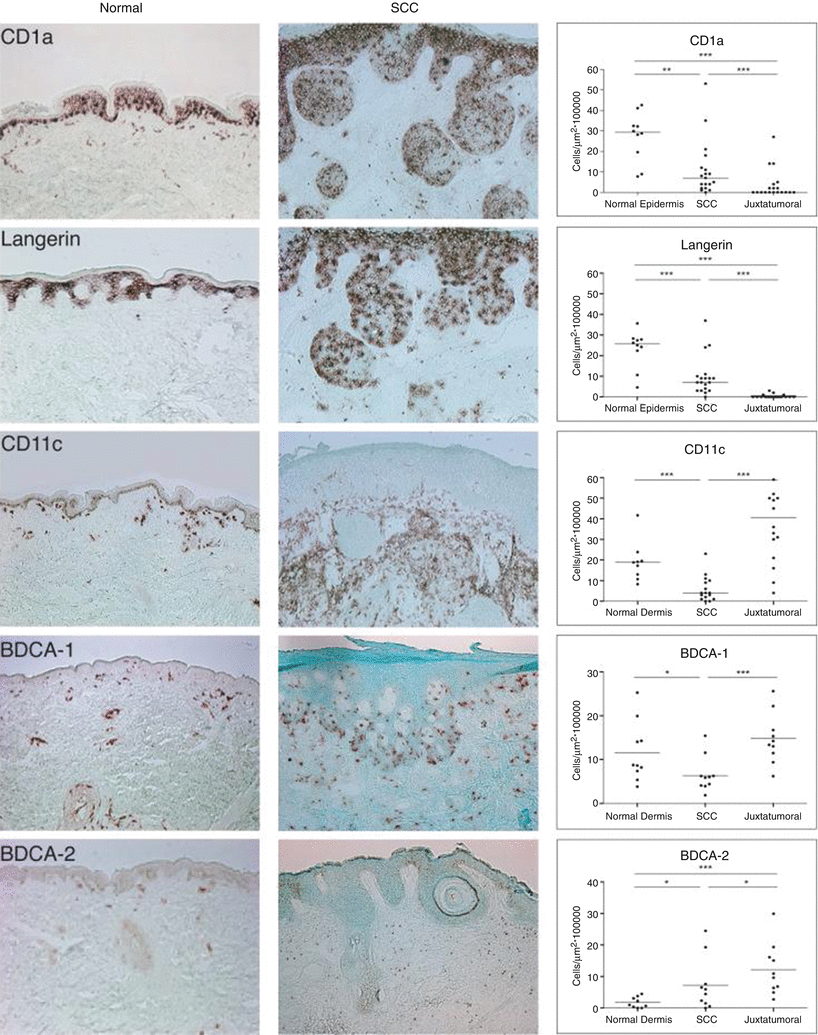
Fig. 42.2
SCC and dendritic cells. Immunohistochemical staining of normal skin and SCC for dendritic cell markers showed that SCC is associated with intratumoral CD1a + Langerin + Langerhans cells, juxtatumoral CD11c + myeloid dendritic cells and BDCA2+ plasmacytoid dendritic cells. (a) CD1a, (b) Langerin, (c) CD11c, (d) BDCA-1, and (e) BDCA-2 cells in normal skin, SCC, and juxtatumoral skin
Additionally, SCC-associated mature myeloid dendritic cells (mDCs) have been shown to be functionally compromised and deficient in their ability to produce IFN-γ and stimulate T-cells, an important indication of the immunosuppressed microenvironment [37]. Even when cultured with mDC-maturing cytokines like IL1b, IL-6,TNF-α, and PGE2, SCC-associated mDCs remained impaired in their T–cell proliferation stimulation. This is despite their phenotypic maturity that is comparable to the mDCs of normal skin. This is thought to be due to the fact that the SCC cytokine milieu has been shown to be composed of immunosuppressive cytokines that suppress myeloid DCs, such as VEGF-A, TGF-β, IL-10, and IL-6 [37].
Langerhans cells (LCs) from SCC seem to be more capable of immune response than their dermal counterparts. LCs from SCC, in contrast to mDCs, have been shown to elicit a type 1 immune response when activated. Moreover, LCs from SCC have been shown to be more powerful stimulators of CD4 and CD8 T cell proliferation than those from normal skin and elicit a more powerful type 1 T-cell response. They also express higher levels of surface markers CD40, CD80, CD83, and CD86 than LCs from normal skin, making them more mature and is an important factor in immune response induction [38].
However, SCC-derived LCs still may have some limitations based on their gene profile. They show a mixed gene profile of upregulated immune activation genes, such as STAT4, IL15, and CD80 as well as upregulated immune tolerance genes, such as CD200 and receptor activator of NF-KB [38]. This mixed activation and tolerance profile is indicative of the tumor environment’s effect on the LCs and more is still to be learned about how this impacts their behavior.
Basal Cell Carcinoma
Basal cell carcinoma (BCC) is the most common human malignancy, with a higher prevalence than all other malignant tumors combined. BCC is a malignant proliferation of keratinocytes from the basal layer of the epidermis. Sun exposure is the most important risk factor for BCC, but other risk factors include age and fair skin. BCC is slow-growing and highly curable, but can be extremely disfiguring if allowed to progress without intervention. BCC has a number of clinical presentations, spanning cystic, ulcerated, nodular, superficial, sclerosing, pigmented, and keratotic variants.
Cytokine and Chemokines in BCC
In BCC, a number of cytokines and chemokines are associated with its avoidance of immunosurveillance and subsequent development. Of the proinflammatory cytokines, IL-6, IL-8, IL-17, IL-22, and CXCL12 have been linked to BCC tumor progression. IL-6 has been shown to increase anti-apoptotic activity within BCC cell lines [39] and promote angiogenesis through the PI3k/Akt pathway, as well as through increasing the expression of the pro-angiogenic cytokine IL-8 [40, 41]. CXCL12 has also been shown to promote angiogenesis by binding to the CXCR4 receptor, which is expressed to a higher degree in more aggressive forms of BCC [42]. CXCL12 also upregulates the activity of matrix metalloprotease 13 (MMP-13), allowing for BCC invasion [43].
IL-17 and 22 have also been shown to increase proliferation and migration of BCC in vitro and tumor progression in vivo, through both their intrinsic signaling pathways and the induction of IL-6 and 8 production [44]. IL-10, an immunosuppressive cytokine, is upregulated in BCC and may contribute to the immunosuppressed microenvironment of the tumor [45]. Its presence has been correlated with lack of HLA-DR, ICAM1, CD40, and CD80 expression, surface markers that aid in immune detection, as well as being correlated with immune cell maturity [46].
IFN-γ is a cytokine that has been shown to stimulate immune responses against cancer. In concordance with a permissively immunosuppressed microenvironment, BCC has decreased expression of IFN-γ receptor, which may play a part in the lack of cell-mediated immune response to BCC [47]. Conversely and logically, IFN-γ is elevated in actively regressing BCC [48]. Interestingly, IL-23, a cytokine which induces IFN-γ production as well as inducing antitumor effects in vitro, has also been shown to be elevated in BCC [49].
Fas ligand (FasL) is an apoptosis-inducing factor that by binding to its receptor on the cell surface, begins the apoptotic cascade of signaling within the cell. It is a member of the tumor necrosis factor family of receptor-ligand binding. When FasL is expressed by cancer cells, it induces the apoptosis of infiltrating lymphocytes, allowing the cancer to evade immune surveillance (Fig. 42.3). Some studies have shown BCC to express FasL, while others have had the opposite results, making the expression of FasL on the part of BCC still debatable [50–52].
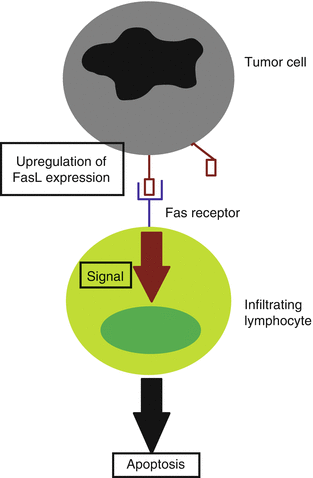
Fig. 42.3
BCC immune evasion through FasL. To evade immune detection, BCC cells upregulate expression of Fas ligand, which binds to the Fas receptor on the infiltrating lymphocyte. This binding inducing a signaling cascade within the lymphocyte, resulting in apoptosis
In general, BCC is associated with a Th2 dominant microenvironment, which is the immune response correlated with immune tolerance. It is capable of significantly inhibiting the Th1 anti-tumor immune response. The Th2 environment is shown with increased expression of IL-4, IL-10, and CCL22, a chemokine responsible for regulatory T cell chemotaxis. However, the BCC microenvironment has also been shown to have an increased expression of interferon-associated genes and IL-23 expression, favoring a Th1 microenvironment [49]. This conflicting microenvironment, shown by the immunostimulatory and immunsuppressive cytokines in the BCC tumor milieu, is consistent with a dynamic state within the immune microenvironment.
Immune Cells and BCC
Dendritic cells, as mentioned earlier, are key in cancer immune surveillance and often have altered behavior in the unique microenvironment of the tumor. In the skin, they exist mainly as epidermal Langerhans cells (LCs) and dermal myeloid DCs. The presence and density of DCs in BCC is controversial, as studies have shown conflicting results. Some studies have shown a decrease and even absence of mature LCs, particularly in tumors of the face, and immature myeloid DCs were found to be present [49, 53], similar to the dendritic cell profile seen in SCC. This decrease in LC density in BCC is linked to increased aggressive behavior on the part of the tumor [54]. Additionally, in BCC cells, the depletion of macrophages and LCs resulted in enhanced tumor progression [55].
Other studies have shown an increased density of LCs within the BCC lesion, as well as in adjacent epithelium [56, 57]. An increase LC density within the BCC lesion is particularly associated with smaller tumor size and tumor location to the face [58]. One explanation for the contradiction in presence or absence of DCs in BCC that has been suggested is that the density of DCs in BCC changes over time, with an initial reduction allowing initiation and subsequent host response increasing DC density [59]. This is also consistent with the dynamic microenvironment suggested by the mixed cytokine profile of BCC.
Regulatory T cells (Tregs) are immune suppressive T cells, important in self-tolerance and immune homeostasis. Created in the thymus, they normally comprise 5–10 % of the CD4 T cells in the periphery. They can also be induced in the periphery from naïve T cells [60]. Both induced and natural Tregs express the transcription factor forkhead box P3 (FOXP3), an important controller of suppressor protein expression and used to identify Tregs [61]. These cells have been found to surround BCC, which is consistent with the increased expression of the chemokine CCL22 (Fig. 42.4). This surrounding of BCC by Tregs can contribute to the immune evasion of the tumor, possibly through the impaired maturation of DCs [49].
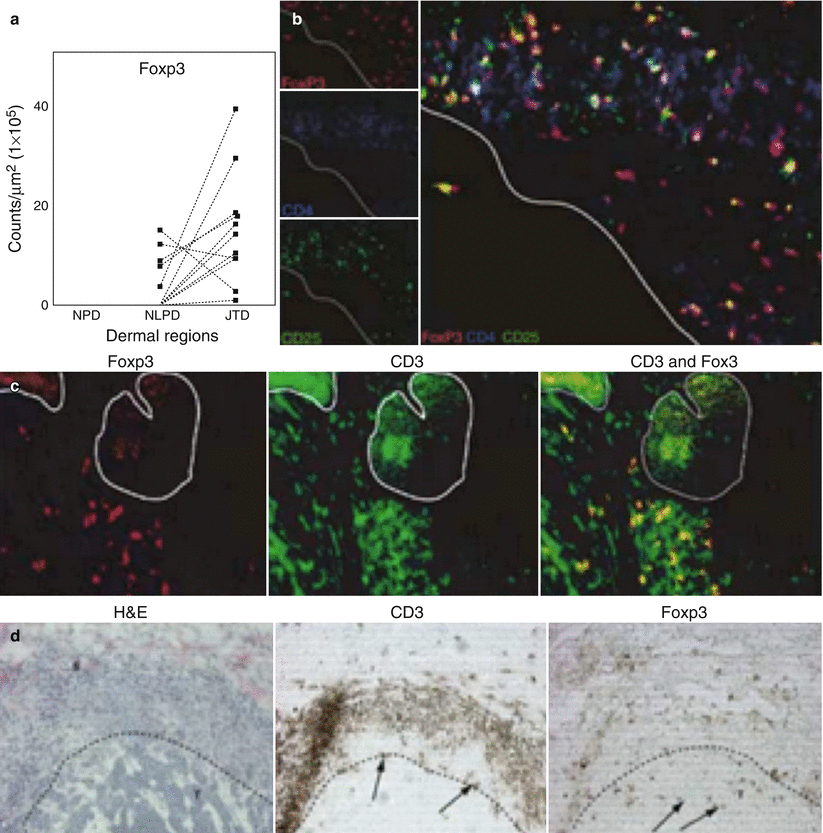
Fig. 42.4
BCC and regulatory T cells. (a) FoxP3+ cells were present in the “pseudocapsule” of inflammatory cells surrounding BCC. (b) Double label immunofluorescence confirmed that FoxP3+ cells expressed CD3. (c) Triple label immunofluorescence showing CD4+CD25+Foxp3+ cells associated with BCC. (d) Cell counts show increased numbers of Foxp3+ cells in juxtatumoral dermis (JTD) versus non-lesional papillary dermis (NLPD) versus normal papillary dermis (NPD)
Mast cells are also thought to contribute to the immunosuppressed microenvironment of BCC. They have been shown to accumulate at the periphery of BCC, especially in aggressive BCC tumors [62, 63]. Additionally, they participate in matrix degradation through the release of proteases, which allow tumor spread and contribute to tumor angiogenesis as a source of VEGF [64, 65], similar to the TAMS of SCC.
Imiquimod
With all the cytokines and immune cells contributing to an immunosuppressed BCC microenvironment, immunomodulatory therapy options were a logical next step to explore. Imiquimod, one of the most successful therapies, is a topical treatment for BCC that has been shown to induce apoptosis in BCC [66]. It is an immune modifier and, more specifically, a Toll-like-receptor-7 (TLR-7) agonist, which induces multiple cytokines, stimulating an innate and adaptive cell-mediated immune response [67].
Imiquimod treatment results in various immune cells invading in stages. The tumor is initially infiltrated by CD4 T cells, followed by a massive intratumoral and peritumoral infiltration of macrophages, as well as activated DCs [68]. The plasmacytoid DCs are recruited by imiquimod’s stimulation of Th1 cytokines, including IL-12. Imiquimod treatment efficacy has been linked to pretreatment DC density in the tumor, with a greater efficiency being shown with a higher density of pretreatment DCs [69]. Imiquimod also induces IFN-α, which in turn induces cell surface expression of FasR on the tumor cells, causing apoptosis of the tumor cells through the CD95 receptor ligand interaction [70–72].
Immunosuppression and Transplant-Associated NMSC
As evidenced by the previous points, the immune system plays a crucial role in the development and progression of non-melanoma skin cancer (NMSC). The most important risk factor for non-melanoma skin cancers is UV radiation. In addition to their mutagen properties, UVA and UVB have been shown to be immunosuppressive in humans. Studies have shown that UVA irradiation suppresses memory immunity to the seven antigens in the delayed-type hypersensitivity test Merieux [73, 74]. Additionally, irradiation with half the UVA in minimal erythema dose (MED), the amount of sunlight necessary to cause a sunburn, suppressed recall immunity to nickel [75]. In animal studies, lymphocytes from UV irradiated mice were unable to prevent malignant formation on UV-irradiated skin grafts in unirradiated mice [76]. Transplanted tumor cell lines, including SCC, which would be immunologically rejected in immunologically competent mice, were able to grow in UV immunosuppressed mice.
Another example of immunosuppression leading to increased risk of NMSC, as well as more aggressive behavior by the NMSC, is the occurrence of these cancers in patients with HIV infection. Immunosuppressed HIV-positive patients can develop rapidly growing cutaneous SCCs at a young age, with an increased risk of recurrence and metastasis [77].
The most obvious and important evidence linking immunosuppression and NMSC is seen in immunosuppressed organ transplant recipients (OTR), with SCC incidence being 60 to 100 times greater in this population than in the age-matched immunocompetent population [78]. The OTR population also has a greater likelihood of multiple skin cancers at presentation (Fig. 42.5) [79].
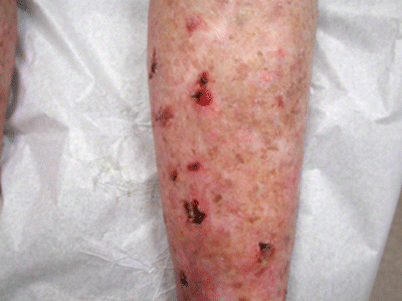
Fig. 42.5
Transplant-associated NMSC. Transplant recipient on long term immune suppression with multiple agents including calcineurin inhibitors presenting with multiple SCCs
Furthermore, SCC in OTR is more aggressive, with a higher probability of recurrence and metastasis [80]. The risk of SCC in OTR seems to be directly proportional to the level of immunosuppression, with lower numbers of CD4 cells found in OTRs with NMSC versus OTRs without NMSC [81, 82].
This altered incidence and behavior of SCC in immunosuppressed patients can be attributed to the altered immune microenvironment. With regards to the modified microenvironment, transplant associated SCC (TSCC) has increased populations of immune cells that are immunosuppressive and produce pro-tumoral cytokines, as well as decreased populations of immune cells that initiate a Th1 (cell–mediated immunity) antitumor immune response [83].
The immunosuppressed TSCC immune microenvironment is altered in its T-cell populations and ratios. More specifically, TSCC has been shown to have increased IL-22-producing CD8+ cytotoxic T cells compared to immune competent SCC, as well as increased expression of IL22 receptors [84]. IL-22 is a pro-inflammatory cytokine typically involved in wound healing, which causes activation of genes involved in cell cycle progression and the prevention of apoptosis [85]. IL-22 has been shown to have pro-tumoral activity in multiple cancers. IL-22 has been shown to drive SCC proliferation in culture in a dose-dependent manner, most dramatically under starvation conditions [84]. This is in concert with the idea that IL-22 may drive SCC proliferation within the tumor, where there are high metabolic demands and diminished enrichment.
Additionally, the CD8+ IFN-γ producing T cells in TSCC are decreased compared to SCC, which is associated with aggressive tumor behavior and increased metastasis [86]. TSCC also has an increased proportion of regulatory T cells (Tregs), which has been correlated with a poor prognosis in other carcinomas, such as breast and gastric (Fig. 42.6) [87, 88]. Tregs are important in preventing autoimmunity, but may suppress beneficial antitumor activity and aid in immune evasion [88–90].
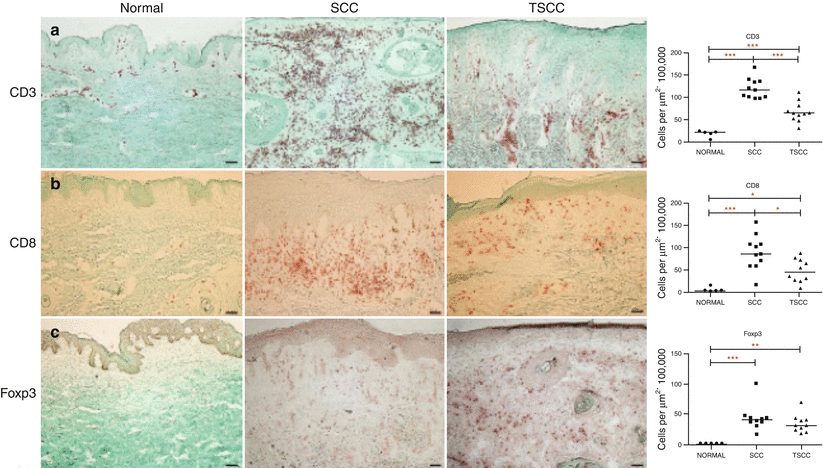
Fig. 42.6
T cell populations in SCC and TSCC. Immunohistochemical staining of normal skin, SCC and TSCC for T cell markers showing the increased amounts of CD3(a), CD8(b) and Foxp3(c) T cells in SCC and TSCC as compared to normal skin. Additionally, the ratio of Foxp3+ Tregs to cytotoxic CD8 T cells was increased in TSCC as compared to SCC, showing a tumor permissive environment in TSCC
TSCC has also been shown to have decreased CD4+ helper T (Th) cells infiltrating the tumor and reduced mRNA for IL17A. IL17A is a Th17-specific cytokine that is involved in inflammation and the recruitment of the innate immune system [91, 92]. The decreased IL17A favors graft tolerance but may weaken the host’s anti-tumor response against the TSCC [93–96].
The skin adjacent to TSCC in OTRs has been shown to be Th2 immune response weighted, with decreased IFN-γ levels [83]. The Th2 gene expression is correlated with transplant tolerance and long-term allograft survival [97]. However, as mentioned earlier, the Th2 response is capable of significantly inhibiting the Th1 response, which is the anti-tumor immune response. This allows for the increased aggressiveness and recurrence rates of SCC in OTRs [83].
These changes in the immune microenvironment suggest a compromised inflammatory response in the SCC of OTRs. This alters the behavior and prognosis of TSCC but is a necessary component of immunosuppression for graft tolerance. Though a minimization of immunosuppression in heart and kidney transplant recipients showed reduction in the development of new SCC at 5 years [98], this reduction is not always feasible. A change in immunosuppression regimen should be considered, with calcineurin inhibitors being linked to higher incidences of cutaneous carcinomas, and sirolimus or everolimus being a better choice with respect to cutaneous tumorigenesis [99]. Lastly, the current knowledge of the immune microenvironment of TSCC opens new avenues of study, with cytokines like IL-22 being possible targets in future TSCC prevention and treatment.
Melanoma
Melanoma incidence rates have been increasing over that last three decades, and greater than 75,000 new cases are projected to be diagnosed in 2014, with over 9500 deaths estimated to occur. Although melanoma is only 4 % of all skin cancers, it accounts for greater that 80 % of skin cancer deaths. About a quarter of melanoma patients will experience recurrence and advanced stage disease [2].
The Immunogenicity of Melanoma
Melanoma is considered an extremely immunogenic tumor, with the capability of triggering host immunologic response. The immunogenicity is seen in the immune cell infiltrates that are often seen in melanoma tumors, as well as the relatively high rate of spontaneous regression with concurrent vitiligo. Many melanoma-specific antigens have been identified in triggering a T-cell immune response. Additionally, a number of immune therapies, such as IFN-α and IL-2, have been shown to be effective in patients, as discussed in the chapter 51 [100, 101].
Despite the established immunogenicity of the tumor, there are a number of immune suppressive mechanisms demonstrated to occur in melanoma, such as impaired antigen-presenting cell maturation, T cell anergy, the induction and recruitment of Tregs, and myeloid-derived suppressor cells [100, 102]. Thus, boosting or stimulating an immune response against melanoma has been and is still a promising avenue for therapy, and its exploration has resulted in some success with regards to disease-free and overall survival [103].
BRAF
In approximately half of human melanomas, activating mutations in the protein kinase BRAF allow the tumor cells to survive and proliferate [2]. BRAF is a proto-oncogene that is part of the RAF kinase family of growth signal transduction. This has been shown to be through the MEK/ERK pathway, which is important in cell division and differentiation. The most common mutation is the V600E BRAF, where a valine at position 600 is replaced by a glutamic acid. This mutated BRAF also has been linked to a deactivated AMPK, or AMP activated protein kinase, which inhibits cell growth in low energy states [104]. With AMPK deactivated, the melanoma cells can then grow despite lack of nutrients.
T Cell Anergy
Melanoma has multiple mechanisms for inhibiting the activation, proliferation and effector status of T cells. This primarily includes stimulating receptors on T cells, including those for PD-L1 and the B7-H4, both members of the same family of co-stimulatory molecules [101].
PD1 is an immunoinhibitory receptor on T cells that plays a crucial role in melanoma’s immune escape. The PD1 ligand (PD-L1 or B7H1) is a member of the B family of costimulatory molecules that provides either an inhibitory or stimulatory secondary signal to T cells primarily binding HLA molecules on tumor cells. More specifically, the B7 family has been shown to control the effector phase of T-cell responses [105]. Specific tumors, such as prostate and renal cell carcinoma, have been shown to express B7 co-stimulatory molecules, which are involved in their escape from immunosurveillance [106].
PD-L1 has been found to be expressed by melanoma tumor cells. When the PD-L1 on the melanoma cell binds to the PD1 receptor on T cells, it causes an inhibition of T cell proliferation, survival and effector functions, such as cytotoxicity and cytokine release. It also induces apoptosis of tumor-specific T cells and promotes differentiation of CD4 T cells into regulatory T cells. Lastly, it increases the resistance of tumor cells to cytotoxic T cell attack [105]. Increased PD-L1 expression by melanoma is negatively correlated with survival (Fig. 42.7) [107].
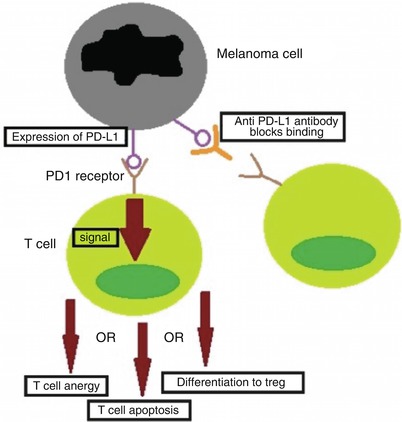
Fig. 42.7
Melanoma immune evasion through PD-L1. One method of immune evasion exhibited by melanoma is the expression of PD-L1 on its surface, which binds to the PD1 receptor on the surface of T cells. The binding of PD-L1 to its receptor causes intracellular signaling, leading to several different immunoinhibitory outcomes: T-cell anergy, apoptosis or differentiation of the T cell into a regulatory T cell (Treg). One of the immune therapies being explored is an anti-PD-L1 antibody, such as nivolumab, which blocks the PD-L1 on the melanoma cells from binding and inhibiting T cells
This pathway is therefore an important therapeutic target, with anti-PD-L1 antibodies, such as nivolumab, tested as immunotherapeutic agents, either alone or in combination with other immune therapies. These have shown some success, but await further testing and demonstration of clinical effectiveness.
Another member of the B family of co-stimulatory molecules, B7-H4, has also been indicated in immune evasion of melanoma. Melanoma tumor cells have been found to express B7-H4, and its presence was shown to also have an inhibitory effect on T-cell cytokine production, in particular IFN-γ, TNF-α and IL-2 [101]. These cytokines are all integral to the T-cell mediated anti-tumor response. As mentioned above, high levels of PD-L1, or B7-H1, have been shown to correlate with reduced patient survival in melanoma. B7-H4 has also been associated with poor patient outcomes, with lower expression levels correlated with better survival [107].
Regulatory T Cells and Melanoma
Tregs, as mentioned earlier, are a dominant mechanism of tumor immune escape. Tregs are overrepresented in the peripheral blood of patients with melanoma and are found in the tumor microenvironment and affected lymph nodes as well [108]. The percent of Tregs infiltrating the melanoma lesions has been shown by some studies to negatively correlate with survival [109]. An even better predictor of survival is the ratio of CD8 T cells to Tregs in the tumor microenvironment [110].
Tregs are thought to accumulate in the melanoma tumor microenvironment through a number of mechanisms. As mentioned earlier, Tregs are both being induced by PD-L1 expressing melanomas and selectively surviving, as melanomas expressing PD-L1 are causing apoptosis in effector T cells. Secondly, chemokine secretion and integrin ligand expression of the melanoma tumor cells attracts Tregs from the periphery. Thirdly, immunosuppressive cytokines locally secreted by melanoma, such as TGF-β and IL-10, can induce and expand Treg populations [100].
Tregs’ immunosuppressive capabilities are enhanced through molecular mechanisms, such as melanoma’s expression of indoleamine 2,3 dioxygenase (IDO), IDO is an immunomodulatory enzyme which allows tumor cell immune escape through the depletion of tryptophan [111].
As Tregs are clearly an important part of melanoma’s immune microenvironment and its ability to progress despite melanoma’s immunogenicity, the removal or suppression of Tregs would seem to be a target ripe for therapeutic intervention. Despite many promising studies, clinical efficacy has not yet lived up to its potential, with agents based on IL-2 suppression (a crucial cytokine for Treg activation and proliferation) and FOXP3 vaccination not consistently showing improvement. Other emerging therapies that modulate Tregs, such as CTLA-4 blocking agents (CTLA-4 serves as an inhibitory molecule constitutively expressed by Tregs) and PD-1 blocking agents, have shown some success and need to be explored further [100].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


