Fig. 37.1
Schematic for granuloma formation. Monocyte/macrophage (Mo/Mac) interaction with an antigen provokes an inflammatory response. Under influence of various cytokines and chemokines, some Mo/Macs undergo cell fusion to form multinucleated giant cells (MGCs). Once formed, MGCs secrete steady-state chemokines, attracting more Mo/Macs. Other activated Mo/Macs enlarge and differentiate into epithelioid histiocytes. Aggregation results in granulomas. Activation by tumor necrosis factor-a (TNF-α) and interferon-g (IFN-γ) is important in granuloma formation and maintenance
Recently, it was demonstrated that during cell differentiation into MGC, high amounts of chemokines were induced in Mo/Mac [18]. However, once the MGC becomefully differentiated, the expression of the chemokines such as CCL2 and CXCL10 become constitutive and can not be further upregulated by exposure to Mycobacterium tuberculosis. This suggests two distinct phases in MGC formation. First, in acute infection, the development of MGC induces rapid recruitment of Mo/Mac via chemokine release. In the second phase, the differentiated MGC stimulates a steady influx of Mo/Mac, presumably towards the development of granulomas, via a steady state of chemokines. The connection between granulomatous infections and chemokines is supported by reports demonstrating high amounts of chemokines in lung specimens of patients with tuberculosis [19, 20].
Granuloma Formation
Presence of granulomas may represent an intact immune protective process, an exaggerated reactive process, and/or an attempt at restoring homeostasis. The experimental model of granuloma formation traditionally relies on a murine model of persistent infection, usually with Mycobacterium species, to examine the interaction of pathogen with Mo/Mac. Research aimed at unraveling the inciting signals has implicated a role for TLRs, the pro-inflammatory cytokine TNF-α, and T-helper 1 (Th1) cytokines IL-2 and IFN-γ [21, 22].
The elicitation of the host’s Mo/Mac TLRs response in the M. tuberculosis model is complex because both pro-inflammatory and anti-inflammatory cytokines have been reported following M. tuberculosis exposure [23]. It is unclear if this dichotomy represents an immune evasion strategy employed by the pathogen or a balancing act by the host in an attempt to control inflammatory responses. Surprisingly, the susceptibility to M. tuberculosis was low in numerous TLRs knockout (TLR2, TLR4, and TLR6) mouse models. However, the myeloid differentiation factor 88 (MyD88) knockout mice displayed high susceptibility to disease thus demonstrating the importance of TLRs in host defense against mycobacteria since MyD88 functions an adaptor molecule used by many TLRs following ligand activation. Studies using mycobacteria and other pathogens have demonstrated the importance of MyD88 in granuloma formation via IFN-γ induction [24–27].
More than a decade earlier, Flynn and colleagues reported that IFN-γ knockout mice infected with M. tuberculosis develop disseminated tuberculosis. They demonstrated that disease susceptibility followed granuloma necrosis due to the lack of macrophages activating signals needed for granuloma maintenance [28, 29]. Th1 cells play a role in both the formation and maintenance of granulomas by inducing and sustaining Mo/Mac recruitment. Similarly, mice deficient in TNF-α succumb to disseminated tuberculosis infection. In the absence of TNF-α, the granulomas formed are structurally disorganized leading to eventual necrosis [30, 31].
Matrix metalloproteinase (MMP), specifically MMP-12, also known as macrophage metalloelastase, has recently been reported to be abundantly expressed in granulomas [32]. Although not directly related to granuloma formation via macrophage activation but rather macrophage migration, MMP-12 has been shown in vivo to be important in macrophage penetration of basement membrane by digesting a variety of stromal substrates [33, 34]. Furthermore, surveys of several different human granulomatous skin disorders, including sarcoidosis, necrobiosis lipoidica diabeticorum, and granuloma annulare demonstrate that MMP-12 is abundantly expressed in co-localization with CD68+ macrophages [35].
While the murine model provides a useful mammalian in vivo experimental tool, there are limitations. For example, cross strain murine infection with Mycobacterium tuberculosis produces multi-bacillary non-caseating granulomas, contrasting the usual course in human disease [36]. Furthermore, study of early events in granuloma formation, namely Mo/Mac recruitment, modification, and aggregation is limited by static ex vivo histological examination of tissue in this model.
Recent development of non-mammalian granuloma models have helped broadened our understanding of granuloma formation. This is possible through examination of host interaction with Mycobacterium marinum in species phylogenetically distinct from humans such as Dictyostelium, Drosophila, and zebrafish [37]. While extrapolation into human disease pathogenesis may not be immediately apparent, the observations suggest that pathogen recognition via pathogen associated molecule patterns (PAMPs) and macrophage aggregation may be evolutionary conserved processes. The Dictyostelium is a single cell amoeba that has similarities to the human phagocytic histiocyte. Drosophila is a known model of innate immunity. By utilizing the transparent zebrafish embryos, researchers have directly visualized migration and aggregation of macrophages, the hallmark of granuloma formation, following infection with M. marinum [38]. Furthermore, at the embryonic stage, zebrafish lack circulating lymphocytes, suggesting that granuloma formation can be initiated in the absence of an adaptive immune contribution.
Granulomatous Disease
Except for infectious and foreign body granulomatosus, the factors determining the non-infectious granulomatous diseases are largely unknown. It likely results from the interplay between genetic susceptibility and environmental factors. Understanding the reaction patterns as well as the cellular and soluble mediators involved will be informative for understanding the etiopathogenesis of granulomatous diseases (Fig. 37.2). While this group of diseases involves infiltration and aggregation of immune cells, this does not necessitate that these disorders are immunologically mediated. Histiocytic aggregation may be seen in reactive processes.
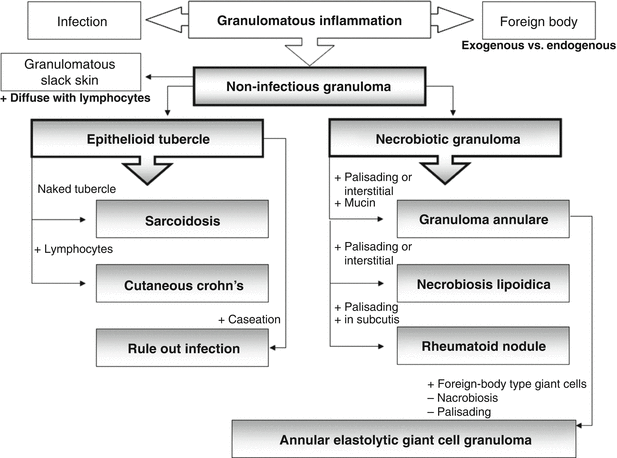
Fig. 37.2
An approach to the histologic diagnosis of granulomas
Sarcoidosis is the most studied non-tuberculid granulomatous disease because of its affect on multiple organ systems and associated mortality and morbidity. Understanding the underlying molecular and cellular processes of this prototypic granulomatous disease may help elucidate the pathogenesis of other non-infectious granulomatous disorders.
Other diseases include cutaneous Crohn’s disease, which is a spectrum of cutaneous manifestations associated with the inflammatory gastrointestinal disease, not all of which are granulomatous. The group of necrobiotic granulomas including granuloma annulare, necrobiosis lipoidica, and rheumatoid nodule share some common histological features but no consistent correlations with a single underlying systemic disease. Except for vasculitis and potential ulceration in the latter entity, the cutaneous conditions are largely benign. Annular elastolytic giant cell granuloma is a rare disease that is considered by some to be a variant of granuloma annulare, but with histologically distinct features. Similarly, it has a fairly benign course and may be self-limiting. Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis are two more recently classified granulomatous diseases that shares histologic features with granuloma annulare and necrobiosis lipoidica, though not much is known about their etiopathogenesis.
Non-infectious Epithelioid Granulomas
Sarcoidosis
Clinical Manifestations
Sarcoidosis is a multisystem disease characterized by non-caseating granulomas of unknown etiology [39]. The most commonly affected organ is the lung. Involvement of the skin is seen in up to one-third of patients, and may be the only clinical sign of the disease. Although the cutaneous lesions usually appear at onset of disease, it can occur at any time. Other organs affected include the liver, spleen, eyes, kidneys, glands, and less commonly, the central nervous system, heart, and musculoskeletal system. Several syndromes have been described in sarcoidosis depending on the constellation of symptoms and involved organs that include: Heerfordt-Waldenstrom syndrome (fever, parotid gland enlargement, anterior uveitis, and facial nerve palsy); Mikulicz’s syndrome (infiltration of the parotid, submandibular, lacrimal, and sublingual glands) [40]; and Lofgren syndrome (fever, erythema nodosum, polyarthralgias, and bilateral hilar adenopathy). The latter is associated with a benign self-remitting course [41]. In general, while the mortality rate associated with severe pulmonary disease is low in sarcoidosis, 10–30 % of patients develop chronic debilitating disease.
Sarcoidosis affects all races worldwide with varying incidence. In the United States, African Americans have a 3.8-fold increased risk compared to whites, affecting women more than men [42]. The disease tends to be more chronic and fatal in African Americans [119].
Several studies have established that the skin disease does not correlate with prognosis or the extent of visceral involvement [42, 43]. However, there is correlation with erythema nodosum, usually a self-limiting condition seen most frequently in young women on initial presentation, with acute disease and large plaques with chronic disease [42, 44, 45]. Recent retrospective analysis of a small cohort of patients with the subcutaneous form of sarcoidosis suggests that this variant may be a subset associated with systemic disease [46].
The most common skin manifestations include non-painful reddish-brown papules and plaques often symmetrically involving the face, lips, neck, trunk, or upper extremities [47, 131]. Typically lesions lack scale or ulceration, though some lesions may develop these features. Cutaneous sarcoid is a great mimic and can have diverse clinical presentations. A variety of uncommon manifestions include an ichthyosiform, annular, angiolupoid (large telangiectatic), psoriasiform, or subcutaneous (Darier-Roussy) appearance. Atrophy can be seen in plaques. Scalp lesions have varying amounts of scales and may result in alopecia. Various non-specific nail and mucosal changes can also be seen.
Another manifestation of sarcoidosis is lupus pernio, indurated and violaceous papules and plaques on the nose, cheeks, and ears [48]. Ulceration can be seen in these lesions and can lead to scarring. Nasal alar lesions can extend into the nasal vestibule and nasal floor and is associated with granulomas in the upper respiratory tract and lungs in the majority of patients [49, 50]. The digits and toes may have sausage-shaped swelling due to underlying cystic lesions of the phalanges [51].
Histopathology
Langerhans, granulomas seen in sarcoidosis have the characteristic appearance of focal collections of epithelioid histiocytes associated with absent or sparse ring of lymphocytes composed of T and B lymphocytes. The term “naked tubercle” refers to a common observation of a granuloma devoid of lymphocytes and plasma cells. Multinucleated giant cells may be present, usually of the Langhans type, and these giant cells may contain asteroid and Schaumann bodies. There is typically no central caseation; however, there may be fibrinoid changes secondary to deposition of immunoglobulins, complement, and fibrinogen in up to 10 % of cases. The presence of caseation should prompt a search for infectious causes.
Both TNF-α and IFN-γ have been associated with granuloma formation [52, 53]. It is known that prolonged activation by these cytokines usually leads to apoptosis but the characteristic granulomas in sarcoidosis patients are non-caseating. It is unclear why there is the absence of apoptosis in the sarcoid granulomas. Recent studies have associated upregulated expression of an IFN-γ induced anti-apoptotic molecule p21Waf1, a cdk inhibitor, in sarcoidosis patients [54].
Pathogenesis
Numerous hypotheses have been put forth on the etiology of sarcoidosis, including infectious, autoimmune, and environmental. The identity of a causative antigen responsible for granuloma formation remains uncertain. The etiology and pathogenesis of sarcoidosis is complicated by highly varied disease presentation ranging from single organ to multisystem involvement, the lack of specific symptomology, and the waxing and waning nature of the disease. Both the clinical and tissue diagnosis of the disease require exclusion of other conditions such as mycobacterial or deep fungal infections, Wegener’s granulomatosus, and malignancy.
A historic test for the diagnosis of sarcoidosis is the Kveim-Siltzbach skin test [55–57]. This was performed by injecting a patient with a suspension of sarcoid spleen material intradermally into the skin of a suspected patient. The formation of a non-caseating granuloma observed histologically 4 weeks later at the site of injection indicates a positive test. Studies of T cells at the Kveim-Siltzbach reaction sites have demonstrated an oligoclonal population of CD4+ T cells which argues for sarcoidosis as an antigen driven disease [58]. This is supported by findings of similar oligoclonality, with a dominant V beta bias, in sarcoid lung T cells [59].
Infectious Etiology
Despite investigations for viral and mycobacterial causes of sarcoidosis, no consistency has been found. Human herpesvirus-8 (HHV-8) was postulated to be associated with sarcoidosis but has been met with many reports of negative findings in sarcoidosis patients worldwide [60–63]. Mycobacterial DNA sequences have been found in various tissues in some sarcoidosis patients, while others report negative findings [64–67]. To date, no mycobacteria have been successfully cultured [68].
Recent reports have identified various mycobacterial components as potential pathogenic antigens in sarcoidosis. Serum from patients with sarcoidosis were found to have antibodies to Mycobacterium tuberculosis katG, M. tuberculosis heat shock protein 70, and Mycobacterium tuberculosis mycolyl transferase antigen 85A [58, 126, 127].
Environmental Exposure
Evidence for the role of environmental exposure in the pathogenesis of sarcoidosis comes from reports of higher incidence of disease in certain occupations, such as firefighters and aircraft carrier personnel [69, 70]. A recent study by Izbicki et al. noted an increased incidence among New York City Fire Department workers involved in the 2001 World Trade Center emergency [125]. Such disease clusters indirectly implicate an environmental etiology [71]. In a recent multi-center case control study, researchers did not find a single proximate cause. This group did report positive associations with insecticides, an agricultural environment, and microbial bioaerosols such as mold and mildew [72]. Interestingly, a negative association with cigarette smoking was found in the study, consistent with previous reports [73, 74].
Genetic Susceptibility
Familial predisposition of sarcoidosis is well-known: having a first-degree relative with the disease confers a five-times increased likelihood of having the disease. There are also reports of sibling discordance [84]. Several studies have reported disease susceptibility with HLA-1, HLA-B8, HLA-DRB1, HLA-DRB3, and HLA-DQB1 alleles [75, 118]. No definitive HLA genes have been uniquely established in African American sarcoidosis patients, a group identified to have 3.8-times increased annual incidence in the United States. Recent linkage studies suggest that more than one gene may be involved in disease susceptibility in African Americans [86]. A recent study by Valentonyte et al. reported an association of the butyrophilin-like 2 (BTNL2) gene on chromosome 6p with sarcoidosis but its precise function in sarcoidosis is still unknown [121].
CARD15/NOD2 has been linked to Blau syndrome, a granulomatous disease affecting the eyes, joints, and skin [76]. CARD15, expressed by mononuclear phagocytes, encodes NOD2 which recognizes a component of bacterial peptidoglycan [77]. The gene has also been associated with inflammatory bowel disease [78]. One report found an association of mutations in CARD15/NOD2 with early-onset childhood sarcoidosis, a distinct type of sarcoidosis in children younger than 4 years of age characterized by eye, joint, and skin involvement [79]. Attempts to find an association of CARD15/NOD2 in adult sarcoidosis have demonstrated no relationship [75].
Despite several proposals of correlative serum markers, including serum amyloid A and C-reactive protein, no consistent correlations have been demonstrated. Even serum angiotensin converting enzyme (ACE), initially promising, has not been a consistent marker of disease activity [80, 81]. Other markers currently being examined include macrophage inflammatory protein 1 (MIP-1) and vascular endothelial growth factor (VEGF) [82, 83].
Immune Regulation
There are numerous lines of research indicating that the involved lymphocytes are of the Th1 phenotype, producing cytokines such as IL-2, IFN-γ, and enhanced TNF-α [87, 88]. Findings of hypergammaglobulinemia in sarcoidosis patients leads to the question of B cell involvement in disease pathogenesis. It is now believed that the hypergammaglobulinemia is secondary to IL-2 and IFN-γ stimulation of B cells [89, 90]. Adding to the puzzle of sarcoidosis is that despite activation of networks of pro-inflammatory cytokines and robust recruitment of cells, there is often an associated state of either complete or partial anergy [91, 92]. This has been demonstrated by a lack of response to the tuberculin skin test or decreased sensitization to agents like the contact sensitizer dinitrochlorobenzene (DNCB) seen in up to two-thirds of sarcoidosis patients. Explanations offered for the observation of decreased delayed-type hypersensitivity was due to peripheral lymphopenia from sequestering and compartmentalization of T cells into granulomas. Recent data suggests that sarcoidosis patients have an unusually high number of innate T regulatory cells (Tregs), with constitutive CD25bright/CD4+ cells, both in the peripheral circulation and at the periphery of granulomas [93]. Tregs from sarcoidosis patients demonstrated similar capabilities of suppressing responder cell proliferation as compared to controls. The Tregs from sarcoidosis patients inhibit IL-2 but not TNF-α or IFN-γ production by responder cells compared to normal controls in which all three cytokines were completely inhibited. Since TNF-α is associated with sarcoidosis and granuloma formation, these findings may explain the concurrence of cell activation with a global state of anergy.
Th17, a CD4+ effector T cell population, has been linked to chronic inflammatory diseases involving Th1 cells including psoriasis, rheumatoid arthritis, and inflammatory bowel disease [117]. Recent studies have shown the presence of Th17 in the lung and peripheral blood of patients with active sarcoidosis [116]. Presence of these cells likely play a role in the chronic inflammatory state found in sarcoidosis [117].
It is known that 8–21 % of patients with common variable immunodeficiency (CVID) develop a granulomatous disease resembling sarcoidosis [94]. Since CVID is not a single entity but rather a heterogeneous syndrome, a retrospective study correlating patients with granulomatous disease and specific immunologic derangement may provide some clues in the pathogenesis of sarcoidosis. Some CVID patients develop hyperproliferation of CD4+ cells, yet some have increased apoptosis of CD4+ cells. Sixty percent of patients have a diminished response to T cell receptor stimulation and expression of CD25, the receptors for IL-2.
Treatment
Many individuals require no treatment, though the decision to treat depends on the site and severity of organ involvement [122]. There are numerous anecdotal and case series that discuss the treatment of sarcoidosis but few clinical trials. The mainstay therapy includes corticosteroids. Other commonly used agents include minocycline (particularly for cutaneous sarcoidosis), anti-malarials, methotrexate, and TNF-alpha agents [95, 122]. Paradoxically, several reports have noted the appearance of cutaneous sarcoid lesions while on TNF-alpha therapy for other diseases [123, 124].
Cutaneous Crohn’s Disease
Crohn’s Disease (CD) is characterized by segmental granulomatous inflammation of the intestinal tract. Cutaneous manifestations of the disease occur in 14–44 % of patients, and include CD-specific, reactive, and associated conditions. CD-specific lesions involve the skin by the same mechanism as the GI tract and include fissures and fistulae, oral Crohn’s disease, and metastatic Crohn’s disease. The latter is a rare entity that denotes a cutaneous lesion distant from extension or fistulae formation from oral, anal, or ostomy sites. Reactive lesions involve the skin by distinct pathogenic mechanisms and include erythema nodosum, pyoderma gangrenosum, Sweet syndrome, and polyarteritis nodosa [96, 128].
The cutaneous manifestations of Crohn’s Disease, including metastatic Crohn’s disease, have inconsistent correlation with internal disease activity. Classically, extra-genital metastatic Crohn’s disease presents with dusky erythematous plaques that may develop into ulcers. Histopathologic examination reveals a sarcoid-like epithelial granuloma, usually with a notable lymphocytic infiltrate and occasionally with necrobiosis [97, 98]. The inflammatory infiltrate can often surround dermal blood vessels, termed granulomatous perivasculitis [98].
A recent study attempted to elucidate the etiology of cutaneous Crohn’s by examining the gastrointestinal and corresponding skin specimens in patients with Crohn’s and cutaneous Crohn’s disease for bacterial 16s rRNA. They report no bacterial 16S rRNA, examined by in situ real time vs reverse transcriptase polymerase chain reaction (RT-PCR), in skin lesions whilst being present in gastrointestinal biopsies, suggesting that bacterial dissemination may not be involved in cutaneous lesions. Reactivity to bacterial products may play a role in the cutaneous manifestation of Crohn’s [99].
As discussed earlier, polymorphisms in CARD15/NOD2 have been associated with Crohn’s disease but, the mechanism leading to predisposition of disease is unclear [100]. One hypothesis is that mutations in CARD15 leads to a defect in the acute inflammatory response to intestinal bacteria, leading to allowance of materials to breach the gut barrier. The subsequent response to the bacterial products leads to a granulomatous reaction [101].
Necrobiotic Granuloma
The group of necrobiotic granulomas consists of granuloma annulare (GA), necrobiosis lipoidica, and rheumatoid nodules. The etiology of these diseases is unknown. The common histological reaction pattern is palisading granulomas with areas of altered or degenerated connective tissue. Variations such as interstitial granulomatous inflammation can be see in GA and necrobiosis lipoidica, as well as interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis, two recently categorized granulomatous diseases.
Granuloma Annulare
GA is a benign disorder limited to the skin. There have been many proposed inciting factors such as trauma, insect bites, and viral infection. A delayed-type hypersensitivity reaction to an unknown antigen may be the formative event; this hypothesis is supported by a study showing T-cell subpopulations in histologic specimens of GA lesions [129]. The association with systemic disease such as diabetes mellitus has been inconsistent. Atypical variants of GA have been described in patients with HIV/AIDS and lymphoma [102, 103]. The localized variant is most commonly seen, occurring as annular or arcuate plaques on the hands and arms [104]. It can also involve the extremities and trunk. Other variants include papular, generalized, perforating, subcutaneous, and patch forms of the disease. On histology, the recognizable pattern is of palisaded epithelioid histiocytes with a central acellular area of pallor, increased mucin, and altered collagen and elastic fibers. The most common pattern is the infiltrative or interstitial pattern where histiocytes are interspersed between collagen with subtle alteration of the fibers.
The entity annular elastolytic giant cell granuloma is regarded by some as a variant of GA. Lesions are also asymptomatic. On histology there are foreign body type giant cells without a palisading granulomatous inflammation. There is usually no altered collagen. An elastin stain shows loss of elastic fibers in granulomatous areas.
Rheumatoid Nodules
Rheumatoid nodules can be seen in the clinical setting of adult-type polyarticular rheumatoid arthritis and is a rare feature of rheumatic fever [105]. History and context of presentation will aid in the diagnosis. The etiology of rheumatoid arthritis (RA) remains unknown. Disease susceptibility is strongly associated with class II region genes, HLA-DRB1 [106–108].
Clinically, rheumatoid nodules present as firm papules or nodules along extensor surfaces of joints. Histology reveals a palisading layer of histiocytes and granulation tissue surrounding a central zone of fibrin [130].
There is high suspicion that RA is mediated by autoantibodies, specifically by a group that recognizes citrullinated proteins, including the antiperinuclear factor, antikeratin antibodies, and antifilaggrin antibodies [109]. These antibodies are commonly found in the sera and synovial fluid of RA patients [110]. In a small study of 26 patients, citrullinated proteins were observed in 70 % of the rheumatoid nodules [111].
Citrullination, a post-translation modification process, has been demonstrated to increase peptide and MHC affinity, suggesting that it can modulate immune responses [112]. The enzyme mediating citrullination is peptidylarginine deaminase, found in inflammatory cells including neutrophils, monocytes, and macrophages [113]. Hence, it is tempting to speculate a relation of the Mo/Mac and granulomatous reaction in the skin with citrullination as a potential source of “autoantigens”. Recently, T cells recognizing the specific modification of antigens by citrullination was described [114]. It remains to be determined if they are the pathogenic mediators of RA.
Necrobiosis Lipoidica
Necrobiosis lipoidica “diabeticorum” (NLD) was originally described in patients with diabetes but demonstration of non-diabetic patients with the condition have lead to reconsideration of its name. The disease has a characteristic presentation of asymptomatic yellow-brown plaques involving the pretibial areas. The lesions over time become atrophic with development of telangiectasias. Ulceration can develop but the disease is generally benign. On histology the “cake layers” of granulomatous inflammation and parallel degenerated collagen is typically seen. The pattern can be palisaded or interstitial.
The etiology of NLD is unknown. Twenty years ago Ullman and Dahl suggested that the disease may be secondary to vasculitis based on findings of IgM and complement C3 deposition in blood vessels of affected skin [115]. Recently, NLD skin sections were shown to stain for GLI-1, a transcription factor in the hedgehog signaling pathway. The significance remains to be seen.
Closing Remarks
Traditionally the Mo/Mac is simply thought of as phagocytes. That frame of reference prompted investigations with the goal to identify causal pathogens in granulomatous diseases. Macrophages are very plastic cells found in most tissues and are responsive to their microenvironment. New understanding of the diverse homeostatic and regulatory functions of Mo/Mac should broaden our understanding of this inflammatory response.
Furthermore, diseases with overactive and chronic persistence of Mo/Mac and granulomatous inflammation can be the potential target of liposomes, specific uptake by Mo/Mac, or via targeting their unique receptors such as anti-Fc©RII receptor (CD 32). However, the degree to which macrophages play a role in promoting lesions and their persistence is not fully understood in some of these diseases. Agents that limit Mo/Mac response may play an important role in understanding the pathophysiology of these dermatoses.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


