The future of melanoma research is promising. Specific mechanisms leading to oncogenic transformation in melanoma development have been identified, and are likely to produce new targets for melanoma therapy. Also, advances in melanoma research will result from melanoma investigators co-opting approaches used to study other malignancies in which progress has been made more rapidly. Systematic roadblocks limiting advances in melanoma research relative to other malignancies are being addressed in a formal manner. The public and public officials are increasingly becoming aware of the need for more dedicated efforts to address the challenges of research on this malignancy.
Although there have been no recently developed, widely implemented therapeutic advances in melanoma in increasing the survival of patients with advanced disease, considerable advances have been made within the past decade highlighting the molecular, cellular, and genetic determinants of malignant melanoma, which have promoted a more detailed understanding of melanoma development and behavior to suggest new therapeutic approaches. High-throughput sequencing efforts of targeted regions of the cancer genome have unexpectedly yielded important findings in the somatic genetics of melanoma, leading to the identification of newly recognized molecular targets. Future comprehensive DNA sequencing efforts using melanoma genetic material should reveal additional characteristic genetic lesions associated with precise clinical subtypes of melanoma as well as their frequencies of occurrence. These findings will suggest novel therapeutic targets. Comparative genome hybridization (CGH) has been used to reveal aberrations of the melanoma genome correlating with the activation of specific cell cycle and signaling genes. In addition to providing more insights into therapeutic mechanisms, these findings will enable the diagnosis of indeterminate pigmented lesions to be made with more precision. As a counterpart to future genetic advances, the application of findings about the histone code and DNA methylation to melanoma will reveal which epigenetic modifications occurring in melanoma cells lead to malignant progression and resistance to therapy. Future findings relating epigenetic markers to tumor formation and prognosis may rival the importance of genetic markers in the future.
Oncogene-induced senescence, regarded as a cellular mechanism protecting against malignant progression following genetic alterations predisposing to cancer, seems to account for the growth arrest of melanocytic nevi despite the presence of an oncogenic mutation in these lesions. Understanding its determinants in melanocytes may be important for devising strategies to revert malignant lesions to a more benign state. Initial findings describing melanoma stem cells, examples of cancer stem cells that have been identified previously in hematologic and nonhematologic malignancies, may lead eventually to the identification of a highly specific subpopulation of melanoma tumor cells, responsible for tumor self-renewal during metastasis, which could be targeted with appropriate agents. Systems biology, a merging of computational techniques and dynamic analyses enabling prediction of the complex effects of changes in cellular states, also represents a technique that could be harnessed to increase the understanding of malignant melanoma. Traditional melanoma immunotherapy has recently incorporated creative introduction of conventional chemotherapy with the use of tumor-infiltrating lymphocytes and gene therapy to boost the success rate of this cell-based therapeutic approach. The need for access to melanoma tumor tissue, primary and metastatic, has been a roadblock to progress in melanoma research, and initiatives undertaken by the National Cancer Institute and other organizations have addressed this in a formal way.
This review concentrates on describing 5 areas in which future advances are likely to impact on the ability to understand, prognosticate, and treat human melanoma: (1) germline mutations, (2) somatic mutations and DNA sequencing, (3) CGH, (4) oncogene-induced senescence, and (5) melanoma stem cells.
Inherited mutations and melanoma susceptibility
The description of hereditary germline mutations accounting for increased melanoma susceptibility not only has had important implications for patient management but also has revealed important determinants, stimulating additional discoveries in melanocyte and melanoma cell biology. The gene CDKN2A at chromosome 9p21, encoding the p16 and p14 tumor suppressor proteins, is the most frequently mutated gene in hereditary melanoma syndrome. Identification of CDKN2A as a major melanoma susceptibility gene led to recognition of the importance of p16 loss or inactivation in the development of sporadic melanoma and melanocyte immortalization. This identification has also resulted in the successful development of mouse models to study melanoma experimentally.
In contrast to the high penetrance of CDKN2A mutations for familial melanoma, another melanoma susceptibility gene that has been identified has low penetrance, and is present widely throughout certain populations of European continental ancestry. This gene is MC1R , encoding a G-protein coupled receptor, the type 1 melanocortin receptor, transducing intracellular signals on binding by its ligand, melanocortin 1 or α-melanocyte stimulating hormone (α-MSH), that facilitates the production of eumelanin (dark or brown-black melanin), inside the melanocyte. The MC1R gene was identified as the “red hair gene,” and certain variants of the gene are associated both with a red-haired, freckled phenotype and an increased susceptibility to melanoma (and nonmelanoma skin cancer) development.
The International Melanoma Genetics Consortium (GenoMEL; www.genomel.org ) was formed to evaluate further the risk of melanoma and other cancers in families with variations in known melanoma susceptibility genes, to identify additional susceptibility genes, and to explore gene-environment interactions in the development of melanoma and other relevant malignancies. A recent report from this initiative confirmed the association of CDKN2A mutations with pancreatic cancer ; it also reported that mutations in the p16 component of CDKN2A accounted for only 38% of the total cases in the study, with mutations in the p14 component of CDKN2A and the CDK4 gene, another rare melanoma susceptibility gene, contributing another 3%. Hence, the majority of variations responsible for familial melanoma in these consortium cases have not yet been identified. Although some of these unaccounted cases may be the result of occult CDKN2A mutations in more distant regulatory regions of the gene, it is likely that mutations in currently unrecognized melanoma susceptibility genes account for many of these cases. Identification of these genes, using consortia of affected families in conjunction with laboratory-based analyses of genetic linkage and additional candidate genes, will identify additional genes that, like CDKN2A and CDK4 , yield greater insights into the biology of melanoma development.
Somatic mutations in melanoma and intensive sequencing-based analyses of the melanoma genome
As described earlier, the discovery of inherited mutations in the germline can reveal important determinants of tumor development in individuals with hereditary tumor predisposition syndromes. However, most melanomas occur sporadically, in individuals without a significant family history of melanoma. To identify important molecular factors contributing to the development of sporadic melanoma, a different approach was necessary, one based on examining the DNA of melanoma tissue to discover genetic alterations that determine malignant transformation and cancer development. For example, members of the RAS family of proto-oncogenes, which transmit intracellular signals following the binding of growth factors to their cellular receptors, are frequently mutated in human cancer. An HRAS mutation in a bladder tumor was the first described somatic mutation in human cancer. Mutations in NRAS were described nearly 2 decades ago in human melanoma , suggesting that activation of RAS-dependent intracellular signaling was also an important characteristic of cutaneous melanoma.
An intensive sequencing-based approach to discover cancer-specific alterations in the RAS-RAF-MEK-ERK signaling pathway (mitogen-activated protein or MAP kinase pathway) led to the discovery that mutations in the BRAF gene, encoding the BRAF serine/threonine kinase, were present in 66% of melanomas analyzed in the study. Eighty percent of BRAF mutations in human cancer occur at a specific site in the BRAF coding sequence, resulting in the substitution mutation BRAF V600E in the protein. This discovery has generated much excitement in melanoma research because of the possibility that BRAF might represent a viable target for molecular-based therapy. Since the initial report, a variety of additional analyses have elaborated on these findings. In melanomas, activating mutations in BRAF and NRAS are frequent but mutually exclusive. Mutations in BRAF are most closely associated with melanomas that develop on skin not chronically exposed to sunlight, such as the skin of the trunk, which receives intermittent, but high-intensity, sunlight exposure. Melanomas occurring on chronically sun-exposed skin, such as the skin of the face, as well as acral and mucosal melanomas, have infrequent activating mutations in BRAF. In contrast, activating mutations in NRAS, usually at codon 61, are nearly equally distributed between melanomas occurring on chronically sun-exposed, nonchronically sun-exposed, acral, and mucosal surfaces. Site-specific differences in mutational frequencies suggest that the intensity and amount of ultraviolet exposures results in cellular changes, promoting the development of distinct genetic lesions within melanocytes and ultimately leading to malignant progression.
In the future, expansive DNA sequencing analyses of melanoma specimens will reveal additional mutations that contribute to melanoma development and pathogenicity. An instructive example to consider is the set of mutations that have been described from intensive DNA sequencing of colorectal carcinomas. A targeted sequencing effort, focusing on the sequencing of genes encoding kinases in the phosphatidylinositol 3-kinase (PI3K) signaling pathway, revealed multiple mutations in the PIK3CA gene, encoding the p110α subunit of PI3K, in colorectal carcinomas at high frequency (32%). These mutations were found to promote growth and invasiveness of colorectal cancer cells, confirming their pathogenicity. A more extensive analysis, the sequencing of important catalytic domains of 340 genes encoding cellular serine/threonine kinases, revealed mutations in 8 additional genes in colorectal carcinoma cells, including 3 genes that contribute to the PI3K signaling pathway that were also implicated in the previous study. The complete cancer genome of an individual patient with acute myelogenous leukemia has been sequenced, revealing 8 previously undescribed mutations that are likely to contribute to cancer progression. Similar efforts to explore novel mutations in kinases controlling intracellular signaling pathways as well as other critical sets of genes, such as the matrix metalloproteinases, in melanoma tissues will define currently unrecognized genes that could represent viable therapeutic targets. These efforts will be particularly important because strategies to inhibit BRAF, though frequently mutated and important for governing melanoma cell growth and survival in vitro, have thus far yielded disappointing results. The use of sorafenib, a multikinase inhibitor that inhibits both BRAF and BRAF V600E activity, in melanoma has not been found to be more effective than current therapies in initial clinical trials. Evaluation of additional molecular targets and signaling pathways identified by DNA sequencing-based approaches is urgently needed.
Somatic mutations in melanoma and intensive sequencing-based analyses of the melanoma genome
As described earlier, the discovery of inherited mutations in the germline can reveal important determinants of tumor development in individuals with hereditary tumor predisposition syndromes. However, most melanomas occur sporadically, in individuals without a significant family history of melanoma. To identify important molecular factors contributing to the development of sporadic melanoma, a different approach was necessary, one based on examining the DNA of melanoma tissue to discover genetic alterations that determine malignant transformation and cancer development. For example, members of the RAS family of proto-oncogenes, which transmit intracellular signals following the binding of growth factors to their cellular receptors, are frequently mutated in human cancer. An HRAS mutation in a bladder tumor was the first described somatic mutation in human cancer. Mutations in NRAS were described nearly 2 decades ago in human melanoma , suggesting that activation of RAS-dependent intracellular signaling was also an important characteristic of cutaneous melanoma.
An intensive sequencing-based approach to discover cancer-specific alterations in the RAS-RAF-MEK-ERK signaling pathway (mitogen-activated protein or MAP kinase pathway) led to the discovery that mutations in the BRAF gene, encoding the BRAF serine/threonine kinase, were present in 66% of melanomas analyzed in the study. Eighty percent of BRAF mutations in human cancer occur at a specific site in the BRAF coding sequence, resulting in the substitution mutation BRAF V600E in the protein. This discovery has generated much excitement in melanoma research because of the possibility that BRAF might represent a viable target for molecular-based therapy. Since the initial report, a variety of additional analyses have elaborated on these findings. In melanomas, activating mutations in BRAF and NRAS are frequent but mutually exclusive. Mutations in BRAF are most closely associated with melanomas that develop on skin not chronically exposed to sunlight, such as the skin of the trunk, which receives intermittent, but high-intensity, sunlight exposure. Melanomas occurring on chronically sun-exposed skin, such as the skin of the face, as well as acral and mucosal melanomas, have infrequent activating mutations in BRAF. In contrast, activating mutations in NRAS, usually at codon 61, are nearly equally distributed between melanomas occurring on chronically sun-exposed, nonchronically sun-exposed, acral, and mucosal surfaces. Site-specific differences in mutational frequencies suggest that the intensity and amount of ultraviolet exposures results in cellular changes, promoting the development of distinct genetic lesions within melanocytes and ultimately leading to malignant progression.
In the future, expansive DNA sequencing analyses of melanoma specimens will reveal additional mutations that contribute to melanoma development and pathogenicity. An instructive example to consider is the set of mutations that have been described from intensive DNA sequencing of colorectal carcinomas. A targeted sequencing effort, focusing on the sequencing of genes encoding kinases in the phosphatidylinositol 3-kinase (PI3K) signaling pathway, revealed multiple mutations in the PIK3CA gene, encoding the p110α subunit of PI3K, in colorectal carcinomas at high frequency (32%). These mutations were found to promote growth and invasiveness of colorectal cancer cells, confirming their pathogenicity. A more extensive analysis, the sequencing of important catalytic domains of 340 genes encoding cellular serine/threonine kinases, revealed mutations in 8 additional genes in colorectal carcinoma cells, including 3 genes that contribute to the PI3K signaling pathway that were also implicated in the previous study. The complete cancer genome of an individual patient with acute myelogenous leukemia has been sequenced, revealing 8 previously undescribed mutations that are likely to contribute to cancer progression. Similar efforts to explore novel mutations in kinases controlling intracellular signaling pathways as well as other critical sets of genes, such as the matrix metalloproteinases, in melanoma tissues will define currently unrecognized genes that could represent viable therapeutic targets. These efforts will be particularly important because strategies to inhibit BRAF, though frequently mutated and important for governing melanoma cell growth and survival in vitro, have thus far yielded disappointing results. The use of sorafenib, a multikinase inhibitor that inhibits both BRAF and BRAF V600E activity, in melanoma has not been found to be more effective than current therapies in initial clinical trials. Evaluation of additional molecular targets and signaling pathways identified by DNA sequencing-based approaches is urgently needed.
Oncogene-induced senescence: converting melanoma cells into a dormant state
Oncogene-induced senescence is a cellular response, characterized by a cessation of cell division, resistance to death, and other morphologic and gene expression changes, to the expression of an activated oncogene. Oncogene-induced senescence was first demonstrated as an in vitro response of fibroblasts to the expression of an activated RAS-family oncogene, and is thought to represent a protective response against the development of actual malignancy. Additional genetic and epigenetic changes in the senescent cell are required for malignant progression, and may convert the activity of the oncogene from mediating cell cycle arrest to fostering malignant progression. Cellular mechanisms enforcing oncogene-induced senescence may thus represent viable targets for therapeutic intervention.
In addition to melanomas, BRAF V600E mutations are found at high frequency in melanocytic nevi. The presence of activating mutations in a RAS signaling pathway member suggested that BRAF activation in melanocytes might induce oncogene-induced senescence. In laboratory-based experiments, introduction of BRAF V600E into cultured human melanocytes was found to result in an arrest of cell division and the induction of characteristic markers of cellular senescence. Some of these same markers are expressed in vivo by melanocytic nevi. All of this evidence suggests that melanocytic nevi result from oncogene-induced senescence in human melanocytes, with an arrest in cell proliferation following an initial stimulus from activation of an oncogene, resulting in the stability of this benign neoplasm. This insight leads to a follow-up question: what are mechanisms that maintain a melanocytic nevus in a senescent state in the presence of an activated oncogene which, in melanoma cells, is capable of driving their tumorigenicity?
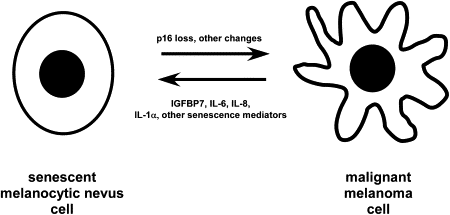
A recent set of studies has defined a set of novel determinants of oncogene-induced senescence that may represent opportunities for future intervention against malignant melanoma. Screening of BRAF V600E -expressing melanocytes led to the identification of insulinlike growth factor binding protein 7 (IGFBP7) as a determinant of oncogene-induced senescence in human melanocytes. In this study, administration of IGFBP7 to mice xenografted with BRAF V600E human melanoma cells induced a significant regression of these tumors. In another study, expression of interleukin (IL)-6 along with associated cytokines IL-8 and IL-1α were found to mediate an oncogene-induced senescence response. A third report implicated expression of the chemokine receptor CXCR2, which binds IL-8 along with other cytokines, for reinforcing cellular senescence responses. As summarized in Fig. 1 , the use of these and other mediators of senescence that have yet to be characterized may be useful in the future for reverting melanoma cells in patients toward a senescentlike state. Although this strategy may not cure melanoma, administration of soluble proteins and cytokines that have the potential for inducing a state of dormancy could be important for achieving long-term control of this malignancy in patients with advanced disease.