Fig. 21.1
Mode of action of the nano-injection system. The intact microcapsule in an anhydrous gel formulation (a) is activated by a hydrophilic drug formulation (b). Water molecules (triangles) and the soluble drug (circles) penetrate the porous wall of the microcapsule, resulting in high internal pressure that causes forcible ejection of the nano-injector and discharge of its microcapsular contents into the skin (c) (Modified from Shaoul et al. (2012))
21.3 Harnessing a Biological System for Drug Delivery
More than 10,000 species of Cnidaria contain nano-injection systems. We were interested in microcapsules with relatively short nano-injectors that would be able to penetrate the SC and enter only the upper part of the viable epidermis. We therefore chose sea anemone microcapsules, which contain smooth nano-injectors 50 μm in length. These microcapsules are isolated, purified, and desiccated, all under sterile conditions, yielding a powder of intact micro-devices whose potential activation is unimpaired (Ayalon et al. 2011), and which, when formulated as an anhydrous gel, can be spread over the skin. Activation occurs only when the gel formulation is hydrated by the addition of a hydrophilic formulation containing the drug (Fig. 21.1). Thus, administration of the drug comprises two steps: first, spreading of the anhydrous gel formulation containing the micro-device system over the skin and, second, application of the hydrophilic drug formulation. Because activation is immediate, the drug is delivered to the epidermis in less than 5 min, and owing to its short and thin nano-sized dimensions, the delivery is virtually noninvasive (see movie in (Ayalon et al. 2011)).
21.4 Delivery of Hydrophilic Compounds
Owing to the characteristics of the skin, hydrophilic compounds are the most difficult molecules to deliver by passive chemical application. The nano-injection system is specifically designed to address this obstacle and hence to facilitate the delivery mainly of hydrophilic compounds. We have previously shown that various compounds, including organic compounds and peptides, can be delivered, via our micro-devices, locally into the skin (Ayalon et al. 2011; Lotan 2005, 2008), or systemically (Shaoul et al. 2012). The unique delivery apparatus is described and illustrated in the next section.
21.4.1 Kinetics of Drug Release
Activation of the anhydrous gel containing the nano-injection system is immediate when combined with a hydrophilic drug. The applied soluble drug is pumped into the microcapsule and through the nano-injector to its target (Fig. 21.1). This process continues until the poly-γ-glutamate driving force of the system is washed out through the nano-injector. As the entire process takes less than a second, we studied the kinetics of the system over a short period (5 min) of exposure to a test drug delivery, after which the drug was removed from the skin by thorough washing. This brief period, although considerably longer than the time required for activation of the system, was chosen because of technical limitations. Using a Franz diffusion cell system, we carried out in vitro measurements of the permeability and kinetics of various test compounds in a sample of full-thickness skin from nude mice. Figure 21.2a demonstrates the delivery kinetics of three hydrophilic drugs (lidocaine hydrocloride, triethanolamine HCl, trolamine salicylate and nicotinamide), all of which are used in topical formulations for various therapeutic purposes such as local anesthesia and arthritic pain as well as in skin cosmetics but which usually necessitate prolonged application to permeate the skin. Exposure of the skin for 5 min to these drugs after applying the gel formulation containing the biological micro-devices resulted, in each case, in the successful permeation through the full-thickness skin of 40 % of the delivered drug as early as 30 min after application and of more than 90 % at 5 h. Control experiments carried out under identical conditions but without the micro-devices resulted in permeation of negligible amounts (Fig. 21.2b). Evidently, therefore, the rapid delivery obtained with the biological micro-devices is not typical for passive topical or transdermal formulations but is rather an outcome of the active mode of penetration via nano-injection.
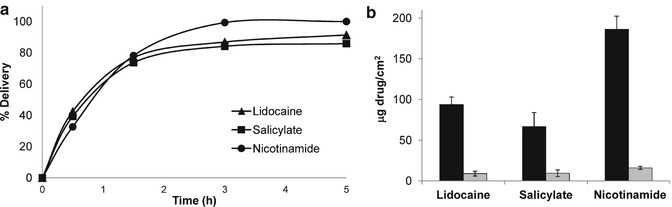
Fig. 21.2
(a) Kinetics of 5 % lidocaine hydrochloride, 5 % trolamine salicylate, and 5 % nicotinamide through the full-thickness skin of nude mice, assessed in a Franz diffusion cell. The microcapsules were applied in gel formulation containing 2 % hydroxypropyl cellulose in absolute ethanol over the skin, and the test drugs were placed over the microcapsule-containing gel for 5 min. The skin was then thoroughly washed and samples were taken between 0.5 and 24 h. (b) Accumulation of delivered compounds indicated in (a), measured 24 h after application of the microcapsule-containing gel (black bar) or of the gel formulation without microcapsules (gray bar). Error bars represent means ± SD
21.4.2 Dose Control in a Peptide Model
The quantity of drug delivered by the nano-injection system can be controlled by regulating three basic parameters: drug concentration, size of application area, and number of microcapsules. Common to most transdermal formulations including the nano-injection system are the first two parameters, resulting in an increased drug delivery as a function of drug concentration and application area size (Ayalon et al. 2011; Lotan 2008). The unique feature of the nano-injection system is that, as with fixed fabricated microneedles, the number of penetration points can be controlled. However, unlike in the case of microneedle use, the concentration of microcapsules in the anhydrous gel formulation that is spread over the skin can be varied.
Our experimental results of topical application of a hydrophilic peptide demonstrate the effect of microcapsule number on drug delivery. Peptides and proteins are relatively limited in their delivery through the SC, and many advanced formulations and physical techniques are being developed for that purpose, only a few of which are currently on the market (Kalluri and Banga 2011). We tested delivery of desmopressin acetate, a 9-amino-acid synthetic replacement for the hormone vasopressin. This peptide is relatively stable and can be traced under the skin (Cormier et al. 2004). Our nano-injection system, in which different numbers of microcapsules in a gel formulation were combined with 5 % desmopressin solution, was applied to the full-thickness skin of nude mice for 5 min. We found that the amount of drug delivered was proportional to the number of microcapsules up to approximately 1 × 106 microcapsules per square centimeter of skin (Fig. 21.3). In other experiments (Ayalon et al. 2011) we found that above this concentration, there was a reduction in the amount of drug delivered, suggesting that the ongoing increase in microcapsule density probably resulted in microcapsules overload, preventing their optimal contact with the skin. The nude mouse skin used in the above experiments is a convenient and readily available model for percutaneous penetration (Simon and Maibach 1998); nevertheless, to verify that successful application is not limited to the anatomy of the mouse skin, we also tested it on the epidermis of the rodent model that most closely resembles human skin, namely, that of the hairless guinea pig (Sueki et al. 2000). The results were similar to those obtained in nude mice, showing that the system is not restricted to a particular model and suggesting that it can be successfully applied to different skin anatomies (Fig. 21.3). Thus, these experiments confirmed that the delivered dose can be controlled by altering microcapsule density and that the system is compatible with peptide drugs. These findings offer promising possibilities for biopharmaceutical drug delivery.
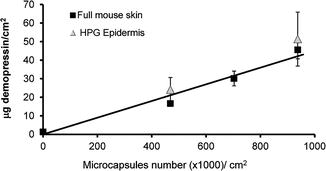
Fig. 21.3
Accumulation of desmopressin acetate delivered to the full-thickness skin of nude mice (black squares) or to hairless guinea pig epidermis (gray triangles), as a function of microcapsule number. Desmopressin (5 % solution) was added to gel formulations containing different numbers of microcapsules per square centimeter of skin or, as a control, to the same gel formulation without microcapsules (0 microcapsules). After exposure to desmopressin for 5 min, the drug was removed from the skin by thorough washing, and the skin samples were left in the diffusion cell for 24 h to allow continuing subcutaneous diffusion of the delivered desmopressin. Error bars represent means ± SD
21.4.3 Systemic Delivery
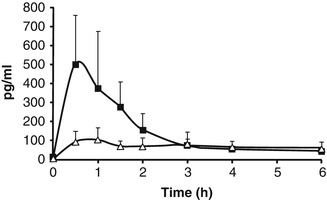
To demonstrate systemic delivery, we used the potent muscarinic receptor antagonist, scopolamine. This naturally occurring alkaloid is one of the most effective single agents used to prevent motion sickness (Spinks and Wasiak 2007) and is also commonly used for the prevention of postoperative nausea and vomiting (Apfel et al. 2010). It was one of the first drugs to be incorporated, into patches for transdermal delivery. However, given the slow incremental diffusion of this drug, the patch has to be applied up to 8 h in advance of need, a major treatment disadvantage (Renner et al. 2005). Because the activity of the micro-injection system is immediate, we expected that its use would enable faster drug delivery resulting in rapidly detectable plasma drug levels. To test this hypothesis, we used a porcine model, known to be particularly reliable for transdermal research in vivo, as comprehensively reviewed by Simon and Maibach (Simon and Maibach 2000). In vivo exposure of the porcine skin to a topical gel consisting of micro-devices combined with 5 % scopolamine hydrobromide was limited to 5 min, after which the sites of application were thoroughly washed, and blood samples were collected. The results showed that the nano-injection system had facilitated rapid accumulation of scopolamine in the plasma, with a time to peak concentration (Tmax) of 30 min and a peak plasma concentration (Cmax) up to five times higher than that of control pigs exposed to similar topical treatment but without micro-devices (Fig. 21.4). Thus, despite its topical formulation, the pharmacokinetic characteristics of the drug in the presence of the micro-devices were comparable to those reported for its subcutaneous injection (Renner et al. 2005).
 Iontophoresis: Basic Principles
Iontophoresis: Basic Principles
 Microporation in Penetration Enhancement
Microporation in Penetration Enhancement
 Combined Use of Ultrasound and Other Physical Methods of Skin Penetration Enhancement
Combined Use of Ultrasound and Other Physical Methods of Skin Penetration Enhancement
 Skin Vaccination Methods: Gene Gun, Jet Injector, Tattoo Vaccine, and Microneedle
Skin Vaccination Methods: Gene Gun, Jet Injector, Tattoo Vaccine, and Microneedle
 Electroporation for Dermal and Transdermal Drug Delivery
Electroporation for Dermal and Transdermal Drug Delivery
 Therapeutic Applications of Electroporation
Therapeutic Applications of Electroporation




Related posts:
Microporation in Penetration Enhancement
Combined Use of Ultrasound and Other Physical Methods of Skin Penetration Enhancement
Skin Vaccination Methods: Gene Gun, Jet Injector, Tattoo Vaccine, and Microneedle
Electroporation for Dermal and Transdermal Drug Delivery
Therapeutic Applications of Electroporation
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
