Commensal bacteria live intimately and in constant dialogue with skin immune cells. Regulating our immune response to these bacteria is critical for skin homeostasis. Using a new murine model to track Staphylococcus epidermidis -specific T cells, we found that colonization during neonatal but not adult life led to S.epidermidis -specific immune tolerance. This tolerance protected against skin inflammation and was mediated by a wave of regulatory T cells entering neonatal skin. These findings provide new insight into how we establish a healthy symbiosis with commensal microbes and highlight avenues for future research to identify novel therapies for inflammatory skin disease.
Key points
- •
Our skin is home to many commensal bacteria that normally do not cause disease.
- •
Regulating our immune response, that is, establishing tolerance, to these commensals is essential to prevent chronic inflammation in skin.
- •
Tolerance to commensal skin bacteria is preferentially established early in life when a unique population of skin regulatory T cells encounters and responds to antigens produced by these bacteria.
- •
Improved understanding of how our skin establishes and maintains tolerance to commensal bacteria may lead to therapeutic approaches to prevent and treat inflammatory skin disease.
Introduction
Commensal Bacteria in Inflammatory Skin Disease
As dermatologists, we routinely diagnose and treat overt cutaneous infections, such as folliculitis or cellulitis, where a discrete pathogen is causative and antibiotics are curative. We are also familiar with autoimmune skin conditions, such as pemphigus or pemphigoid, where an immune response to a self-antigen results in destructive skin inflammation, and treatments aim to limit this via immunosuppression. However, we also care for many patients with inflammatory skin disorders that are neither infectious nor autoimmune by classical definitions. A few examples of these include atopic dermatitis, acne vulgaris, and hidradenitis suppurativa. Although the pathogenesis of these diseases is clearly multifactorial, it is likely that immune responses directed at the cutaneous microbiota help to drive inflammation ( Fig. 1 ).
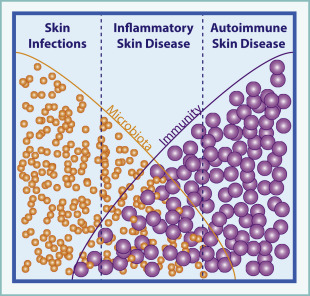
In atopic dermatitis, hereditary defects in skin barrier integrity or host immunity can confer disease susceptibility, perhaps by driving altered responses to skin bacteria. Flares are accompanied by an increase in the cutaneous burden of Staphylococcus aureus and Staphylococcus epidermidis . In acne vulgaris, age of onset coincides with a shift in composition of the skin microbiome. As sebaceous activity increases, the proportion of Propionibacterium acnes on healthy skin increases. The presence of P acnes alone is not sufficient to cause disease, but sequencing of P acnes isolates from acne lesions versus healthy skin has revealed a distinct subset of disease-associated strains. In hidradenitis suppurativa, patients suffer from skin lesions that share many clinical features with infectious furuncles or abscesses. However, microbiological studies of hidradenitis lesions consistently demonstrate altered bacterial communities in which commensal strains from skin or other mucosal body sites predominate over skin pathogens. Thus, in these conditions the presence of a single bacterial strain is not sufficient to initiate disease. Rather, shifts in skin flora composition, accompanied by an altered immune response to these bacteria in susceptible hosts likely trigger pathogenic inflammation.
Present treatment for these inflammatory skin diseases include antibiotics; that is, a sledgehammer to reduce the burden of skin flora and topical or systemic immunosuppressive agents to blunt the resulting immune response. Understanding how our cutaneous immune system regulates inflammation directed against skin microbes will provide additional insight into the pathogenesis of these conditions and may open new opportunities to optimize host–microbe interactions for therapeutic benefit.
Introduction
Commensal Bacteria in Inflammatory Skin Disease
As dermatologists, we routinely diagnose and treat overt cutaneous infections, such as folliculitis or cellulitis, where a discrete pathogen is causative and antibiotics are curative. We are also familiar with autoimmune skin conditions, such as pemphigus or pemphigoid, where an immune response to a self-antigen results in destructive skin inflammation, and treatments aim to limit this via immunosuppression. However, we also care for many patients with inflammatory skin disorders that are neither infectious nor autoimmune by classical definitions. A few examples of these include atopic dermatitis, acne vulgaris, and hidradenitis suppurativa. Although the pathogenesis of these diseases is clearly multifactorial, it is likely that immune responses directed at the cutaneous microbiota help to drive inflammation ( Fig. 1 ).
In atopic dermatitis, hereditary defects in skin barrier integrity or host immunity can confer disease susceptibility, perhaps by driving altered responses to skin bacteria. Flares are accompanied by an increase in the cutaneous burden of Staphylococcus aureus and Staphylococcus epidermidis . In acne vulgaris, age of onset coincides with a shift in composition of the skin microbiome. As sebaceous activity increases, the proportion of Propionibacterium acnes on healthy skin increases. The presence of P acnes alone is not sufficient to cause disease, but sequencing of P acnes isolates from acne lesions versus healthy skin has revealed a distinct subset of disease-associated strains. In hidradenitis suppurativa, patients suffer from skin lesions that share many clinical features with infectious furuncles or abscesses. However, microbiological studies of hidradenitis lesions consistently demonstrate altered bacterial communities in which commensal strains from skin or other mucosal body sites predominate over skin pathogens. Thus, in these conditions the presence of a single bacterial strain is not sufficient to initiate disease. Rather, shifts in skin flora composition, accompanied by an altered immune response to these bacteria in susceptible hosts likely trigger pathogenic inflammation.
Present treatment for these inflammatory skin diseases include antibiotics; that is, a sledgehammer to reduce the burden of skin flora and topical or systemic immunosuppressive agents to blunt the resulting immune response. Understanding how our cutaneous immune system regulates inflammation directed against skin microbes will provide additional insight into the pathogenesis of these conditions and may open new opportunities to optimize host–microbe interactions for therapeutic benefit.
Content
Skin Commensal Bacteria: How Do We Keep the Peace?
Billions of bacteria, viruses, and fungi reside on our skin’s surface and in adenexal structures. Langerhans cells can protrude through tight junctions to capture bacterial antigens on the skin’s surface, and bacterial components have even been identified deep in the dermis. This close proximity enables a constant dialogue between these commensals and our immune system. The presence of bacteria augments the skin’s production of antimicrobial peptides and alters the number and function of skin-resident lymphocytes. Indeed, individual strains of commensal bacteria, such as S epidermidis and P acnes , elicit distinct profiles of cytokine production by skin lymphocytes, demonstrating that the composition of our skin flora can influence the tissue’s immunologic “tone.”
A primary function of our immune system is to protect us from infections by recognizing and responding to microbial antigens. The observation that our immune system is clearly responding to our skin commensal bacteria on an ongoing basis leaves us with a fundamental question that has important implications for normal skin biology and the pathogenesis of inflammatory skin disease. Why do our commensal bacteria not elicit chronic inflammation in healthy skin?
Regulatory T Cells: Our Immunologic Peacekeepers
Our immune system is constantly making decisions about whether and how to respond to antigens it encounters. Most of these antigens are our own “self” antigens. Although many self-reactive T cells are deleted during development in the thymus, others escape and their response must be regulated locally in the tissues where these antigens reside. Regulatory T cells (Tregs), a CD4 + T-cell subset, play a central role in this process of immune regulation or tolerance. As evidence of this, deficiency in the number or function of Tregs leads to autoimmune disease and inflammation in skin and other tissues.
Our commensal microbes are in many ways an extension of our human “self”—not only do we rely on them for critical metabolic functions but, as noted, commensal antigens are pervasive at our body surfaces. Commensal bacteria in our gut have been shown to augment the number and function of Tregs in the intestinal lamina propria, and gut inflammation seen in the absence of Tregs is directed in part toward luminal microbiota. The skin, like the gut, has a significant population of tissue-resident Tregs. However, the role of these Tregs in immune tolerance to skin bacteria was unexplored until recently.
A Good (Immunologic) Relationship Gets off on the Right Foot
The beginning of life represents a critical window of immune maturation in which our immune system is trained to recognize self and non-self. Neonates, especially in prematurity, are more susceptible to certain infections. Previously this was thought to be because of the “immaturity” of their immune systems. Instead, recent evidence suggests that the immune response in this early stage of life is not underdeveloped but rather carefully designed to promote tolerogenic responses. In particular, Tregs generated early in life have a unique propensity to protect tissues from autoimmune attack. Likely this is an adaptive feature to limit potentially damaging immune responses to many new antigens (self and non-self) that the immune system encounters in this developmental window. Colonization by commensal microbiota also occurs at the beginning of life, suggesting that perhaps we educate our immune system to recognize and tolerate commensal microbes at the same time as we learn to tolerate our own antigens.
A New Model to Track Commensal-Specific T Cells and Tolerance
We set out to dissect mechanisms that help us to regulate our adaptive immune response to skin commensal bacteria. T cells have unique surface T-cell receptors, enabling each cell to recognize and respond to a specific antigen. Studying tolerance necessitates isolating just those T cells capable of responding to that antigen and tracking their response in the context of a broad immune repertoire. Tools have not yet been developed to identify and study individual T cells that respond to native antigens made by skin commensal bacteria. Thus, we engineered a skin commensal to express a foreign peptide for which tools are available to track the antigen-specific response ( Fig. 2 ).
We chose to examine the immune response to S epidermidis , a prevalent commensal on human skin that also functions as a commensal in mice, and engineered it to express the foreign peptide, 2W (Epi-2W). A subset of CD4 + T cells in wild-type mice are capable of responding to 2W. These cells can be isolated and studied by flow cytometry using a major histocompatibility complex class II tetramer that binds to the T-cell receptor unique to these cells.
We colonized the skin surface of adult wild-type mice with Epi-2W and examined total inflammation in the skin tissue as well as the 2W-specific (ie, S epidermidis -specific) CD4 + T cells in these animals. After skin colonization, we observed expansion of S epidermidis -specific T cells in both skin-draining lymph nodes and spleen without any accompanying skin inflammation. This suggested that we had successfully created a model of skin commensalism in which we could track a commensal-specific immune response. The robust S epidermidis –specific immune response validated previous lines of evidence that antigens from skin commensals are detected by the immune system even in the setting of an intact physical skin barrier.
A Window of Opportunity: Commensal-Specific Tolerance Is Established in Neonatal Life
We hypothesized that the timing of colonization by a skin commensal might impact the host’s ability to regulate the inflammatory response elicited by this foreign antigen, that is, immune tolerance. To test this, we colonized skin of neonatal or adult mice with Epi-2W and challenged them several weeks later with Epi-2W in the setting of mild barrier disruption alongside naïve age-matched controls. We chose this approach to elucidate commensal-specific immune responses because it recapitulates exposure to commensal antigens in the setting of incidental skin trauma, a mildly inflammatory context during which mechanisms of immune tolerance would need to be active.
Only mice colonized with Epi-2W during neonatal life demonstrated immunologic tolerance to Epi-2W upon challenge, as measured by significantly diminished skin inflammation, reduced skin neutrophils, reduced numbers of S epidermidis –specific effector CD4 + cells in the lymph nodes, and dramatic enrichment of S epidermidis –specific Tregs in both skin and lymph nodes. These results demonstrate that colonization of neonatal but not adult skin results in commensal-specific T-cell tolerance ( Fig. 3 ).
Related posts:
 Basic Science Insights into Clinical Puzzles
Current Status of Dedicator of Cytokinesis-Associated Immunodeficiency
Understanding Inherited Cylindromas
Basic Science Insights into Clinical Puzzles
Current Status of Dedicator of Cytokinesis-Associated Immunodeficiency
Understanding Inherited Cylindromas
 Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
 Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


