Introduction
Disorders of sexual development (DSD)―a less derogatory term than intersex―are a heterogenous group of conditions characterized by aberrant chromosomal, gonadal, and/or anatomic sex with widely varying genotypic and phenotypic manifestations. Sexual differentiation comprises three successive steps: (1) genotypic events determined at conception in regard to chromosomal composition; (2) phenotypic events driven from the chromosomal makeup, which induce gonadal differentiation; and (3) gender identity formation, which can be a lifelong, evolving paradigm predicated on issues such as hormonal induction, external genitalia, and psychologic factors, to name a few. Patients with DSD are generally identified at birth, usually based on physical exam; then a multidisciplinary approach is used to manage these complex issues surgically, psychologically, and socially. These patients are treated by pediatric specialists who are rigorously trained to handle the complex pathophysiology of DSD conditions.
In contrast, transgender patients are genotypically and phenotypically “normal” but have a gender identity that is not congruent with their biologic sex. These patients are usually first evaluated by the physician as adolescents or adults, whereas most DSD patients are plugged into the medical system from a much earlier age. Transgender patients historically have had a much tougher time finding physicians trained in the nuances of their care and only recently have found more holistic and inclusive medical services to meet their needs.
This chapter provides an overview of DSD and the special considerations for these patients. The medical intricacies of DSD and their nuanced treatment is beyond the scope of this chapter. Instead, it delves into a historical overview of sex assignment with special focus on the current guidelines; it concludes by looking at the role of karyotyping in the transgender population.
Embryologic Basis of Sexual Differentiation
An understanding of the embryologic development of the internal and external genitalia is crucial in discussing DSD. We will first discuss the basic steps of normal sexual differentiation so that we can then explain the mechanisms that may alter normal development. Chromosomal gender is determined at the time of conception, as XY chromosomes will lead to male development and XX chromosomes will progress to female development. Early in development, primordial undifferentiated germ cells are arranged in the yolk sac; they will later migrate, become primitive testicles or ovaries, and assist in sexual development.
Two components of the embryo are the precursors to the internal reproductive organs—the paramesonephric (or Müllerian) ducts and the mesonephric (or Wolffian) ducts. Female development is essentially the “default” pathway of development, as the absence of components of the Y chromosome allow for the development of ovaries, fallopian tubes, the uterus, and vagina from the paramesonephric ducts. The so-called testis-determining factor, encoded on the short arm of the Y chromosome (the SRY, or sex-determining region of the Y chromosome) induces growth of the sex cords into primitive testes, where Sertoli cells begin producing Müllerian inhibiting substance (MIS). Without SRY, the sex cords differentiate into ovarian follicles. MIS causes regression of the Müllerian system and stimulates testosterone production from the testicle around the eighth week of gestation, which causes regression of the Müllerian system and stimulates development of the mesonephric duct into the epididymis, vas deferens, and seminal vesicles.
External genitalia remain undifferentiated until the third month of gestation. As shown in Fig. 4.1 , urogenital folds fuse anteriorly to form the genital tubercle and lateral swellings form the labioscrotal folds. Testosterone is converted to dihydrotestosterone (DHT), which acts on the external genitalia to enlarge the tubercle into the penis and fuse the urogenital and labioscrotal folds in the midline.
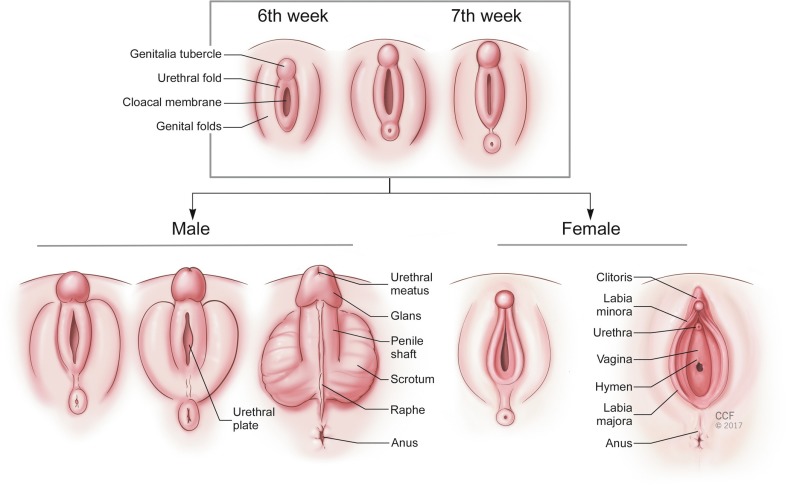
Without androgens, the genital tubercle forms the clitoris and the perineal structures do not fuse in the midline, allowing formation of the vaginal opening. In summary, the production of testosterone and dihydrotestosterone (DHT) is central to the formation of the internal male structures, the involution of the internal female structures, and the development of the male external genitalia. This provides a framework within which most cases of DSD can be understood. There are XX individuals who are virilized as well as XY individuals who are undervirilized due to exposure to androgens or lack of exposure, respectively.
Historical Overview of Sex Assignment at Birth and in Childhood: Current Guidelines
Sex assignment for DSD is a critical medical, social, and legal event that is expected to occur at birth and is often revisited in childhood. Many practitioners argue that this event is of central importance to the care of individuals with DSD. In particular, immediately postpartum, parents of DSD children find it particularly distressing to be uncertain regarding the gender assignment of their child, as gender role determines naming, rearing behaviors, and social interactions with other family members. The onus on the medical and surgical teams is to provide guidance in the sex assignment process, and early incorporation of DSD-specialized physicians is encouraged. Specialists typically include pediatric endocrinologists, pediatric surgeons and urologists, gynecologists, pediatric psychologists, geneticists, and neonatologists.
Historically, sex assignment was primarily driven by phenotypic examination of the newborn, followed by the most current techniques in surgical reconstruction for gender assignment. The construction of female genitalia is more feasible than the creation of male genitalia ; as a result, more DSD patients have been reared as females. Early work to standardize this decision-making process can be traced back to John William Money and colleagues at the Johns Hopkins Hospital in the 1950s. Money, a psychologist by training, conducted a decades-long clinical practice alongside pediatric endocrinologists and surgeons and developed a clear guideline for determining sex, based on the feasibility of surgical reconstruction of the external genitalia. Money states the following in his 1957 manuscript:
Our findings point to the extreme desirability of deciding, with as little diagnostic delay as possible, on the sex of assignment and rearing when a hermaphroditic baby is born. Thereafter, uncompromising adherence is desirable. The chromosomal sex should not be the ultimate criterion, nor should the gonadal sex. By contrast, a great deal of emphasis should be placed on the morphology of the external genitals and the ease with which these organs can be surgically reconstructed to be consistent with the assigned sex.
Money subscribes to the theory of a blank psychologic slate of gender at birth. Implicitly, Money’s research holds two postulates as central to his unifying theory of gender assignment, and these were articulated in a 1997 review of sex assignment at birth. Money’s work was predicated on two fundamental beliefs: (1) that “individuals are psychosexually neutral at birth” and (2) that “healthy psychosexual development is intimately related to the appearance of the genitals.” In effect, Money’s work not only adhered to a standard that guided the sex assignment of DSD individuals but also implied that gender identity was a predominantly psychologic and social construct that was not rooted in biology. These two postulates have been strongly challenged in recent decades, and a more critical determination of sex assignment is now in practice that takes into account a DSD individual’s specific chromosomal, genetic and gondal status in assigning gender at birth.
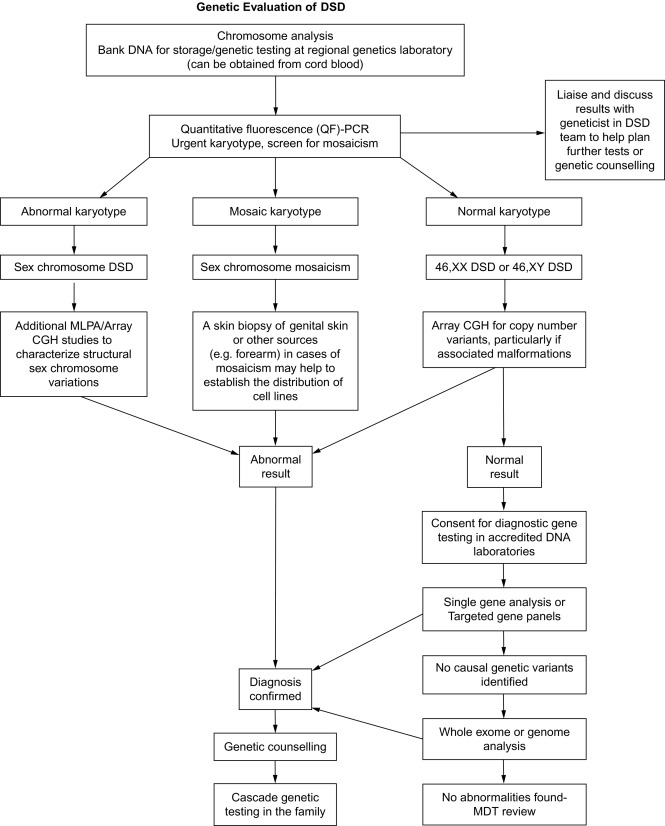
These changed views have led to the development of consensus guidelines to address the evaluation and management of DSD individuals at birth. A central theme of these guidelines is that only a minority (20%) of DSD newborns will have a definitive genetic or molecular diagnosis. Evaluation begins with careful physical examination of the newborn by an experienced clinician to evaluate the external genitalia, including a detailed investigation of the labioscrotal folds and their fusion; the presence or absence of gonads in the abdominal, inguinal, or scrotal position; the size of the phallus and the site of the urinary meatus. These landmarks may not be readily apparent on external examination and thus may require surgical exploration. Prior to any exploration, however, the clinician should follow a well-established algorithm that attempts to specify the chromosomal, molecular, and/or anatomic diagnosis of the DSD. A reformatted algorithm to establish the chromosomal or molecular etiology of each specific DSD is shown in Fig. 4.2 ; it begins with karyotyping to establish the distinction between an overvirilized 46XX-chromosomal female versus an undervirilized 46XY male, or chromosomal mosaicism. Further investigation includes hormonal testing, including 17-OH progesterone, testosterone, gonadotropin-releasing hormone (GnRH), anti-Müllerian hormone, an electrolyte panel, and urinalysis. Abdominal and pelvic ultrasound should be performed to determine the presence or absence of Wollfian or Müllerian structures. Hormonal stimulation tests can be performed as well, including human chorionic gonadotropin (hCG) and adrenocorticotrophic hormone (ACTH) stimulation. Last, specific genetic determination, although rare, as mentioned earlier, can be performed by microarray or full-sequencing technologies of known sex-determining genes in an attempt to identify a specific genetic defect. Notably, other than the physical exam and imaging, all these laboratory tests take more than a week to be completed. This delay can cause additional stress to the family, and, as providers, we should be sensitive to this.
Given the relative frequency of specific etiologies of DSD, the consensus guidelines can help in assigning gender for these conditions. In a 2006, the “Consensus Statement on Management of Intersex Disorders” by Hughes et al. was published, providing guidelines for sex assignment in cases of DSD. These guidelines recommend that 46XX females with congenital adrenal hyperplasia (CAH) and clitoromegaly be reared as females, with surgical correction for the most virilized of cases. Similarly, patients with 46XY with complete gonadal dysgenesis and 46XY patients with complete androgen insensitivity syndrome (CAIS) should also be reared as females. Contrary to previous recommendations, 46XY males with a micropenis are to be reared as males. Similarly, patients with isolated 46XY hypospadias should be reared as males. A particularly interesting DSD example is the 46XY patient with 5-alpha reductase (5-AR) type 2 deficiency. The majority of these individuals identify as males later in life; however, prior to puberty, many are reared as females because of their phenotype. A nuanced view should be taken with these patients, as studies suggest that individuals with 5-AR-type 2 deficiency who have a phallic length greater than 6 cm report satisfactory sexual function as males.
Per the consensus statement, the majority of patients with DSD (including conditions such as 17-beta-hydroxysteroid dehydrogenase [HSD]-3 deficiency, partial androgen insensitivity syndrome [PAIS], incomplete gonadal dysgenesis, cloacal exstrophy, and ovotesticular DSD) do not have specific recommended gender assignments. Instead, anatomic variances, gonadal function after puberty, as well as social and parental influences should be considered when assigning gender to these patients. A comprehensive multidisciplinary evaluation with endocrinologic, pediatric, and urologic providers may assist in the early determination of gender in these patients.
Overview of Disorders of Sexual Development
DSD can be broadly divided into two categories: undervirilization of 46XY males and virilization of 46XX females. The following text considers some of the more common DSDs that providers should be aware of and specifically focuses on issues of gender identity with regard to each condition. More specific medical and technical surgical issues as they pertain to the listed DSDs are beyond the scope of this chapter; the reader is therefore directed to specialized pediatric urologic reconstruction studies.
Undervirilization of 46XY males
Undervirilization of a male with karyotype 46XY can be due to the defective production of testosterone from the testes during the critical period of sexual differentiation, a receptor defect in detecting testosterone, a defect in the conversion of testosterone to other necessary hormones needed for masculinization, or the impaired action or production of MIS.
Disorders of Androgen Synthesis or Action
Androgen Insensitivity Syndrome (Complete or Partial)
Androgen insensitivity syndrome, previously termed “testicular feminization syndrome,” can be divided into complete and partial forms. In CAIS, patients are characterized as having a 46XY karyotype with female external genitalia, bilateral testes, and absence of the Müllerian structures. From a genetic perspective, CAIS is an X-linked condition with an incidence of 1 in 20,000 to 1 in 64,000. Many genetic mutations (e.g., deletions, substitutions, transitions) have been identified in CAIS, but in all cases there is a defect in the gene encoding the androgen receptor, which impairs the target organs’ ability to detect and/or process testosterone. CAIS patients will exhibit normal-appearing external female characteristics with the exception of appropriate secondary pubic and axillary hair. Interestingly, at puberty these patients will have breast development secondary to increased plasma estradiol; as a result, they develop a feminine body habitus. CAIS is one of the DSDs that may not be identified at birth, as these patients phenotypically appear like normal female neonates. However, with the increasing adoption of prenatal genetic screening, more of these patients can be identified at birth. Often, they are diagnosed when they are incidentally found to have testes during inguinal hernia repair surgery or during evaluation for primary amenorrhea in early adolescence. At this age, patients present with diminished pubic and axillary hair and a genital exam that is notable for a blind-ended vagina and absence of a cervix. Imaging will confirm complete absence of the Müllerian structures. Diagnosis of CAIS is further confirmed with karyotype testing, as these patients are always 46XY.
Surgical management of these patients is predicated on the optimal timing of gonadectomy. Historically, patients underwent gonadectomy shortly after birth, when pediatric anesthesia was deemed safe, so as to mitigate the increased risk of testicular malignancy. The downside to early gonadectomy is the postoperative need for exogenous estrogen therapy to induce puberty and the need for yearly dual energy x-ray absorptiometry (DEXA) scans to screen for compromised bone mineral density. Consensus guidelines have recently changed and experts now recommend that the gonadectomy be delayed until after puberty to allow peripheral aromatization of testosterone to estradiol, which would then facilitate appropriate pubertal physiologic changes. Studies have found that the risk of testicular malignancy in CAIS is much lower than previously thought; the incidence is now estimated to be at approximately 2%. Most cases are seminomas and occur in adolescence or later in life. CAIS patients have been thought to have a female gender identity, but recent studies have found that their gender identities are variable, with a higher than expected proportion of patients reporting a nonandrophile sexual orientation.
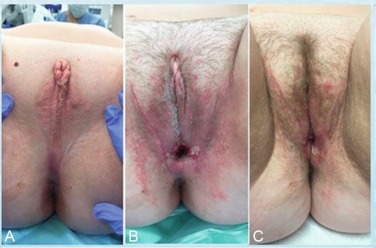
PAIS is a collection of X-linked disorders characterized by a wide array of phenotypic manifestations of incomplete masculinization including the presence of ambiguous genitalia. These patients have a 46XY karyotype but unlike their CAIS counterparts, they are often identified at birth. Classically, patients are found to have some combination of hypospadias, rudimentary Wolffian duct remnants, gynecomastia, and infertility. Hundreds of mutations have been identified in PAIS but most commonly, patients have a reduced number of androgen receptors or decreased binding affinity in these receptors for testosterone. Treatment for patients with PAIS is predicated on the inevitable virilization of the ambiguous genitalia and their subsequent appearance. If raised as male, patients with cryptorchid testes undergo orchidopexy; gynecomastia is addressed and genital reconstruction is performed. Surveillance of the testicles by a urologist is important, as there is an increased risk of testicular malignancy in these patients. Contrary to CAIS patients, patients with PAIS are considered at high risk for testicular malignancy. Therefore in those patients who are raised as female, early gonadectomy is recommended. In these patients, genital reconstruction with procedures such as bowel vaginoplasty with labioplasty is an option, and we report excellent outcomes at our institution ( Fig. 4.3 ) . Last, patients raised as females will need adjunctive hormone therapy starting at puberty. Gender identity in patients with PAIS is an intriguing topic, and there is evidence showing that brain androgen receptors have the same mutation for partial androgen insensitivity as the other end-organ targets. Studies have found that the gender in which a PAIS child is reared often correlates with that individual’s gender identity when he or she is an adult.
Related posts:
 Mental Health Care for the Adult Transgender Patient
Mental Health Care for the Child and Adolescent Transgender Patient
Hormone Treatment for the Adult Transgender Patient
Primary and Preventative Care for Transgender Patients
Gynecologic Care for Transgender Patients
Hysterectomy for the Transgender Man
Mental Health Care for the Adult Transgender Patient
Mental Health Care for the Child and Adolescent Transgender Patient
Hormone Treatment for the Adult Transgender Patient
Primary and Preventative Care for Transgender Patients
Gynecologic Care for Transgender Patients
Hysterectomy for the Transgender Man
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


