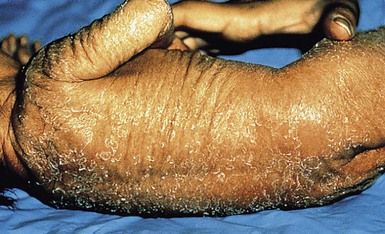
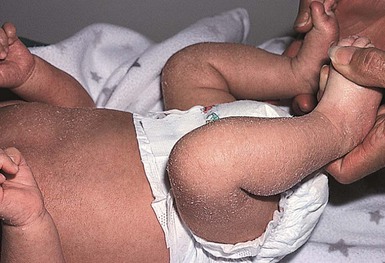
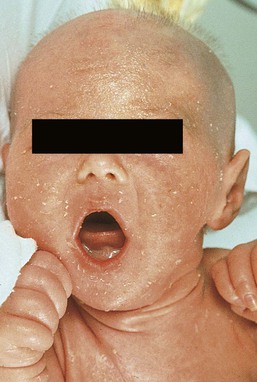
Catherine C. Foley, Amy S. Paller, Alan D. Irvine The term ‘inherited disorders of cornification’ covers a wide range of genetic conditions with molecular defects that preclude the formation of a normal epidermis. The term is usually considered to include entities divided on morphological grounds into ichthyoses, follicular keratoses, and palmoplantar keratodermas. In addition, many inherited disorders usually considered as ectodermal dysplasias have significant defects in epidermal development or differentiation. Several ichthyotic conditions first manifest in the neonatal period, usually as either collodion baby or scaling erythroderma, or more rarely as a harlequin fetus. In some situations, such as harlequin ichthyosis or Netherton syndrome, associated complications are life-threatening. For most of these conditions, therapy during the neonatal and early infantile periods is supportive (Box 19.1), involving frequent application of bland emollients and monitoring for evidence of infection or fluid and electrolyte imbalance. The use of topical medications with keratolytic agents neonatally and during the first 6 months of life is usually unnecessary and risks significant absorption of potentially toxic substances (e.g., lactic acid, salicylic acid). General principles of care for affected infants over 6 months of age include prevention of water loss, emolliating and softening of the stratum corneum. This can be achieved with short, 5 minute baths twice daily, and regular application of emollients. Keratolytic agents, such as urea, lactic acid or salicylic acid compounded with emollients, may be used to remove hyperkeratotic scales. These are not always well tolerated in younger children. Topical corticosteroids may be used for concomitant inflammation, however systemic absorption may be increased in patients with poorly formed cornified layers. Antibacterial washes or bleach baths are beneficial in patients with thick scale, who are at increased risk of cutaneous infection, especially from Staphylococcus and dermatophytes. In patients with suspected dermatophyte infection, skin scrapings should be taken for confirmation prior to local or systemic treatment with antifungal agents. Affected individuals may have impaired sweating due to occlusion of eccrine ducts and care should be taken to avoid overheating. Consider vitamin D3 supplementation in affected children as they may be at increased risk of developing rickets, due to a reluctance to expose their skin to sunlight. In the past 15 years, the molecular bases of the great majority of these disorders have been elucidated, thereby laying the groundwork for confirmatory molecular diagnosis and opening up the possibility of genotype–phenotype correlation and DNA-based prenatal diagnosis for several of the devastating forms of ichthyosis. An international consensus for the classification of inherited ichthyosis was published in 2010.1 Tables 19.1 and 19.2 summarize relevant conditions based on this consensus classification. At present, molecular diagnosis is not available for all forms of ichthyosis and therapy is not yet gene-based. In addition, access to genetic diagnostics is costly and varies from country to country and among different health insurance providers. A wonderful support group is available for all families with a disorder of cornification, the Foundation for Ichthyosis and Related Skin Types (FIRST; www.firstskinfoundation.org). Collodion babies are encased at birth in thickened, shiny, variably erythematous skin that resembles cellophane (Fig. 19.1). The collodion baby (Figs 19.1, 19.2) is the phenotype at birth of several ichthyotic disorders, but variable severities of autosomal recessive congenital ichthyoses (ARCI, non-syndromic) are the eventual phenotype in most patients.2 Others (syndromic forms) include Sjögren–Larsson syndrome, Conradi–Hünermann syndrome, trichothiodystrophy, and neonatal Gaucher disease.3 In 5–6% of collodion babies, normal-appearing skin replaces the collodion membrane, and these babies have the mildest form of ARCI, termed spontaneously healing, self-healing, or self-improving collodion baby.4 Despite the thickening of the stratum corneum, the collodion membrane is actually a poor barrier, which can result in excessive transcutaneous fluid and electrolyte loss with resultant hypernatremic dehydration,5,6 increased metabolic requirements, and temperature instability owing to increased evaporative cooling. Collodion babies are often premature, and the combined skin disorder and prematurity further increase the risk of complications. In addition, numerous cutaneous fissures may be present which, together with the poor skin barrier, increase the risk of the skin being a site of entry for bacteria and subsequent sepsis. Infection may also be difficult to diagnose owing to the intrinsic temperature instability and fluid imbalances associated with the underlying skin condition. Aspiration of squamous material in the amniotic fluid may lead to neonatal pneumonia.7 In addition, the thickening of the skin may restrict movement, making sucking, eye closure, and rarely respiration, difficult. The underlying basis for collodion babies is varied, reflecting the different forms of ichthyosis that present as collodion babies, and these are discussed below. The most common cause is mutations in transglutaminase 1 (TGM1). The self-healing collodion baby phenotype was first shown to be due to mutations in TGM1.8 In two affected siblings, increased hydrostatic pressure significantly reduced the activity of the mutant enzyme, suggesting that this pressure both traps water molecules and locks the mutated enzyme in an inactive trans conformation in utero. After birth, these water molecules are removed and the enzyme is predicted to isomerize back to a partially active cis form, explaining the dramatic improvement of this skin condition.8 Subsequent publications have shown that the ‘self-healing’ variant can reflect underlying mutations in ALOX12B or ALOXE3, in addition to TGM1.9 Several conditions can result in the collodion baby phenotype (Box 19.2). Occasionally, severe cases can be confused with harlequin ichthyosis. Collodion babies should be placed in high-humidity environments to increase hydration, and bland emollients should be applied (see Box 19.1). Electrolytes should be monitored,5 as should fluid intake and output. The membrane usually sloughs during the first month of life (Fig. 19.2). The use of topical keratolytic agents should be avoided in view of the increased potential for toxicity resulting from absorption through the compromised permeability barrier.5 Ichthyosis vulgaris is one of the most common genetic disorders of skin, occurring in approximately 1 in 250 individuals, based on a survey of healthy English schoolchildren.10 In contrast to other forms of ichthyosis, ichthyosis vulgaris does not manifest during the neonatal period. The condition usually appears after 3 months of age as fine, light-colored scales that are larger and coarser on the lower extremities. Palmoplantar markings are accentuated (hyperlinearity). There is an association in some cases with atopic asthma and rhinitis in later life, and ichthyosis vulgaris is associated with a strong risk for atopic dermatitis. Mutations in the filaggrin gene (FLG) have now clearly been shown to underlie ichthyosis vulgaris, leading to decreased to absent expression of filaggrin.11 These mutations are semidominant; heterozygotes exhibit a very mild phenotype with incomplete penetrance, whereas homozygotes or compound heterozygotes show much more severe disease. The mutations show a combined allele frequency of ~4% in Caucasian populations, explaining the high incidence of ichthyosis vulgaris. FLG gene mutations are now well established as the highest genetic risk for atopic dermatitis. In affected boys, ichthyosis vulgaris in the young infant may need to be distinguished from X-linked recessive ichthyosis (see below). In the neonatal period no specific care is necessary. Good skin care with regular emollients and the avoidance of irritants such as detergents is advisable, as these infants tend to have lifelong dry skin and a high incidence of atopic dermatitis.10 Recessive X-linked ichthyosis (RXLI) is a disorder that affects 1 : 6000–1 : 2000 males. The ichthyosis manifests by 3 months of age in 84% of patients, although only 17% show evidence of exaggerated neonatal desquamation and peeling at birth. Extensor surfaces, the preauricular areas, and the sides of the neck are most severely affected by the large, dark, adherent scales (Fig. 19.3). RXLI is regarded as syndromic when accompanied by associated manifestations and non-syndromic when ichthyosis is isolated.1 The absence of steroid sulfatase activity during fetal life also leads to increased fetal production of DHEAS (dehydroepiandrosterone sulfate) and decreased placental estrogen production, which may delay the progression of parturition. Rarely, affected boys have hypogonadism with undescended testes, hypoplasia of the penis and scrotum, and/or failure of normal sexual maturation. The development of testicular cancer has been described in one patient without undescended testes. Approximately 10% of affected boys have a contiguous gene deletion syndrome, a larger deletion which encompasses genes that are contiguous with the steroid sulfatase gene on the terminal short arm of the X chromosome. Deletion of surrounding genes results in mental retardation, hypogonadism, and anosmia (Kallmann syndrome), or a bone dysplasia characterized radiographically by stippled epiphyses (X-linked recessive chondrodysplasia punctata). X-linked ichthyosis results from mutations of the STS gene encoding steroid sulfatase (arylsulfatase C), particularly deletions (90% of patients). In a study assessing the clinical and molecular features in 28 patients with Kallmann syndrome 1, submicroscopic deletions were found at Xp22.3 in four contiguous genes, VCXA, STS, KAL1, and OA1.12 Recessive X-linked ichthyosis (RXLI) in the neonate is not associated with collodion membrane and is therefore distinguishable from other ichthyotic disorders associated with collodion membranes and early skin thickening. Ichthyosis vulgaris is an important differential diagnosis in older male infants and can be distinguished by fluorescent in situ hybridization (FISH) and other genetic analyses. Patients with RXLI, who additionally have a concomitant FLG mutation, have a more severe manifestation of their RXLI. Babies with the rare autosomal recessive disorder, multiple sulfatase deficiency, show scaling typical of RXLI and decreased steroid sulfatase due to a global deficiency of sulfatases. Affected patients also show neurologic abnormalities characteristic of metachromatic leukodystrophy, and features of storage diseases because of the deficiency of several additional sulfatases. In the neonatal period, no specific care is necessary, but patients will generally need lifelong skin care advice and appropriate emollients. RXLI may be detected prenatally. The most common scenario for this is in pregnancies not known to be at risk with an abnormal ‘triple screen’ test that detects decreased maternal estriol levels. RXLI may then be confirmed by FISH, STS (steroid sulfatase), and DHEAS for deletions and/or the demonstration of decreased placental sulfatase activity in amniotic fluid cells and increased DHEAS levels in amniotic fluid. Patients are born with thickening of the skin, including the palms and soles, with generalized prominent follicular keratoses and mild erythema.13,14 The clinical findings have been described as a ‘nutmeg grater’.15 The scalp is hairless, and severe photophobia is noted from birth. The nails may be dystrophic, and follicular pustules may be present. Biopsies show a hyperkeratotic stratum corneum with a thinned dermis. The hair follicles are atrophic and shortened, with abnormal localization of the bulbs to the deep portion of the dermis, rather than a subcutaneous location. There are no normal hair shafts, and sebaceous glands are absent. It is possible that at least two other forms exist in addition to this classic form.16,17 IFAP syndrome is an X-linked disorder caused by functional deficiency of membrane-bound transcription factor protease, site 2 (MBTPS2).20 This results in disturbed differentiation of epidermal structures evoking the triad of ichthyosis follicularis, atrichia and photophobia.20 Female carriers may have linear involvement.21 An autosomal recessive form has also been described. The constellation of clinical signs should make the diagnosis apparent. Systemic retinoids have been used in children as young as 3 years, with reported improvement.22 Most cases of chondrodysplasia punctata are the X-linked dominant Conradi–Hünermann–Happle form. Affected neonates are usually female, because the disorder is considered lethal to male fetuses. However, Conradi syndrome has been described in a few male patients with and without Klinefelter syndrome.23 At birth, patients most commonly have patterned erythroderma with overlying thin to thick psoriasiform scale (Fig. 19.4). In severe cases, generalized ichthyosiform erythroderma with thick scale is seen at birth,24 and later shows typical patterning along Blaschko’s lines with scale desquamation. Involvement may be predominantly unilateral. With advancing age the ichthyosiform erythroderma and stippling improve, leaving finer scaling without underlying erythema and follicular atrophoderma. Cicatricial alopecia occurs as scalp scaling resolves. Psychotropism may be seen (see ‘CHILD syndrome’, below). Extracutaneous features include limb reduction, typically asymmetric, and facies with frontal bossing, saddle nose, and malar hypoplasia. Asymmetric, focal stippled calcifications of the epiphyseal regions are common in childhood but typically disappear in adulthood. Bone defects normally begin soon after birth and during childhood as punctate calcifications, as a result of abnormal calcium deposition during endochondral bone formation. They typically appear in the epiphyses of the long bones, but may also develop in the scapulae, clavicles, sternum, ribs, and spinal column. These lesions usually disappear during adulthood.25 Cataracts usually develop later during childhood, but may be present at birth.23 The underlying molecular basis is mutations in emopamil binding protein (3β-hydroxysteroid-Δ8, Δ7-isomerase), which is involved in cholesterol synthesis (see below).26 Chondrodysplasia punctata can be inherited in both autosomal and X-linked fashion, and may also be the result of an environmental insult, particularly fetal exposure to warfarin.24 The differential diagnosis usually includes two other forms of chondrodysplasia punctata: autosomal recessive rhizomelic chondrodysplasia punctata,24 and X-linked recessive chondrodysplasia punctata with steroid sulfatase deficiency.27 The rhizomelic form is also associated with multiple peroxisomal defects. The ichthyosis occurs in approximately one-third of patients and is poorly described. Affected patients have developmental retardation and tend to die as infants. The X-linked recessive form of chondrodysplasia punctata occurs as a contiguous gene deletion syndrome of Xp, not at the site of Conradi syndrome. The ichthyosis is consistent with recessive X-linked ichthyosis, but stippled epiphyses are associated. Affected infants are deficient in steroid sulfatase activity, and have no peroxisome defects. There is no specific care for the neonate with Conradi syndrome. The term ‘CHILD syndrome’ is an acronym for ‘congenital hemidysplasia with ichthyosiform nevus and limb defects’. The condition occurs almost exclusively in girls, and is presumed to be lethal in affected males. The only case in a boy is thought to represent early postzygotic mosaicism.28 The inflammatory ichthyosiform skin lesion of CHILD syndrome may be present at birth or develop during the first few months of life.28,29 It is characterized by yellow, waxy scaling and is strikingly unilateral, generally with a sharp demarcation at the ventral and dorsal midline regions (Fig. 19.5). Streaks of inflammation and scaling can also follow Blaschko’s lines, with involvement of the apparently unaffected side of the body. Similarly, streaks of normal skin may be interspersed within the area of the CHILD nevus. With increasing age, the skin lesions may improve or clear spontaneously, but thickened erythematous plaques in intertriginous areas tend to persist and be the most severely affected sites (psychotropism).30 The skin lesions of CHILD syndrome nevus can occur without any other abnormalities, but the occurrence of all features of CHILD syndrome in a sibling of a patient with only the CHILD nevus suggests variable expressivity within the spectrum of CHILD syndrome.31 A variable degree of ipsilateral skeletal hypoplasia is an important feature of CHILD syndrome. As with the skin changes, unilaterality is not absolute and slight changes may be present on the contralateral side. Punctate epiphyseal calcifications may be demonstrable by X-ray, but tend to disappear after the first few years of life. Cardiovascular and renal abnormalities are the major visceral problems in CHILD syndrome, although anomalies of other viscera have been described.29 Biopsy of skin lesions shows epidermal acanthosis with marked parakeratosis alternating with orthokeratosis. Basophilic ghost cells of the granular layer are common. The papillary dermis is often filled with histiocytes showing foamy cytoplasm, resulting in the characteristic histopathologic pattern of verrucous xanthoma. Patients with CHILD syndrome have mutations in 3β-hydroxysteroid dehydrogenase, an enzyme in the cholesterol biosynthetic pathway.32 The nevus of CHILD syndrome needs to be distinguished from inflammatory linear verrucous epidermal nevus and linear psoriasis by the histopathologic features and the constellation of other clinical manifestations, if present. CHILD syndrome shares features with Conradi–Hünermann syndrome (see above), a disorder that results from mutation in another gene within the cholesterol biosynthetic pathway; shared characteristics include the prevalence in girls with a presumed X-linked dominant inheritance pattern, ichthyosiform erythroderma, limb reduction defects, stippled epiphyses, and peroxisomal defects. The unilateral nature of the nevus and limb deformities helps to distinguish these conditions. Patients with CHILD syndrome tend to tolerate topical medications poorly, other than bland emollients. However, dramatic improvement has been demonstrated in adults with twice daily application of compounded 2% lovastatin and 2% cholesterol.33 Orthopedic involvement may be needed to manage the limb hypoplasia, including with a prosthesis or even partial amputation. Multisystem care may include cardiology and renal evaluation as needed. Netherton syndrome should be suspected in the neonate with generalized scaling erythroderma, especially if there is failure to thrive (Fig. 19.6).34 Affected infants are often born prematurely and develop the typical ichthyosiform erythroderma in utero or during the first weeks of life. A collodion baby phenotype is not associated. The classic hair shaft abnormality, trichorrhexis invaginata (‘bamboo hairs’, ‘ball-and-socket deformity’), is thought to result from a defect in keratinization of the internal root sheath. Multiple hairs from different areas should be examined, as only 20–50% of hairs may be affected. Although trichorrhexis invaginata may be present in the neonatal period, delayed and sparse hair growth at this time, as well as the easy breakage of these hairs, makes demonstration of the hair defect in the neonatal period difficult. Ichthyosis linearis circumflexa, the characteristic skin change associated with Netherton syndrome, is usually not seen before 2 years of age, but occurs eventually in 70% of patients. It manifests episodically, often lasting for a few weeks and then clearing for weeks or months. The ichthyosiform erythroderma, however, frequently improves with age. The atopic diathesis becomes problematic in two-thirds of patients, with the development of pruritic atopic dermatitis, multiple food allergies, and often accompanying urticaria, angioedema, asthma, and/or anaphylaxis. The failure to thrive is often profound, requiring hospitalization for nutritional support and correction of the hypernatremic dehydration that may be associated. Patients may have diarrhea, and occasionally demonstrate villus atrophy if intestinal biopsy is performed. Most patients have increased levels of IgE, but other laboratory and clinical evidence of immune dysfunction may be present as well. The increased risk of sepsis occurs as a result of both the abnormal skin barrier and the associated immune defects that are only in part owing to malnutrition. The underlying molecular basis for Netherton syndrome is mutations in SPINK5, a serine protease inhibitor.35,36 Lymphoepithelial Kazal-type-related inhibitor (LEKTI), a putative serine protease inhibitor, is strongly expressed in differentiated keratinocytes in normal skin. The lamellar granule system transports and secretes LEKTI earlier than its potential serine protease targets kallikrein 5 and kallikrein 7, thus preventing degradation of desmoglein 1 and premature loss of stratum corneum integrity/cohesion.37,38 Deficiency of LETKI also leads to excessive cleavage of profilaggrin to filaggrin, further affecting differentiation. Netherton syndrome in the neonatal period must be distinguished from several other disorders with extensive scaling erythroderma, failure to thrive, and increased risk of infection, particularly several immunodeficiency disorders, other ichthyoses, and atopic or psoriasiform erythroderma (see Chapter 18). Skin biopsy sections show subacute or chronic seborrheic or psoriasiform dermatitis with spongiosis.39 The stratum corneum is thin and focally parakeratotic, and the granular layer is reduced. Electron microscopic studies have revealed features in skin biopsy specimens that are specific to Netherton syndrome, particularly the premature secretion of lamellar body contents, foci of electron-dense material separating lipid membranes, and disturbed maturation of lamellar membrane structures. Immunohistochemical staining to LETKI, the protein product of SPINK5, is also possible and with increasing availability will become a highly useful diagnostic investigation,40 but is currently not widely available. Treatment of Netherton syndrome is extremely difficult. Despite their pruritic, erythematous skin, patients tend to respond poorly to topical steroids, although some have improved after topical application of calcineurin inhibitors or calcipotriol. The poor cutaneous barrier of patients with Netherton syndrome may result in significant absorption of topically applied agents. For example, high blood levels of tacrolimus have been found following topical application for the associated dermatitis.41 The application of keratolytic agents or administration of systemic retinoids often worsens the severity of the disorder, and their use is inappropriate during the neonatal period. Most patients prefer to use bland, thick emollients as the only therapy throughout life. Collodion membrane and absent hair may be presenting features at birth. Patients display varying degrees of ichthyosis, jaundice, scarring alopecia and hypotrichosis, not necessarily all obvious from birth. Congenital paucity of the bile ducts may be a primary feature leading to hepatomegaly, cholestasis and hepatic fibrosis, without fatty infiltration.42 Hepatic involvement varies from transient cholestasis to liver failure. Mild psychomotor delay and bilateral anterior uveal synechiae have been described along with oligodontia, hypodontia and dysplastic enamel.42,43 This is a rare autosomal recessive disorder caused by homozygous mutations affecting the CLDN1 gene coding for the tight junction component claudin-1, leading to lack of expression of CLDN1 in skin and liver.44 Consider this syndrome in any child born with collodion membrane, alopecia and neonatal cholestasis. Treatment should follow recommendations for collodion babies. In addition, the hepatic manifestations should be evaluated and managed by specialists.
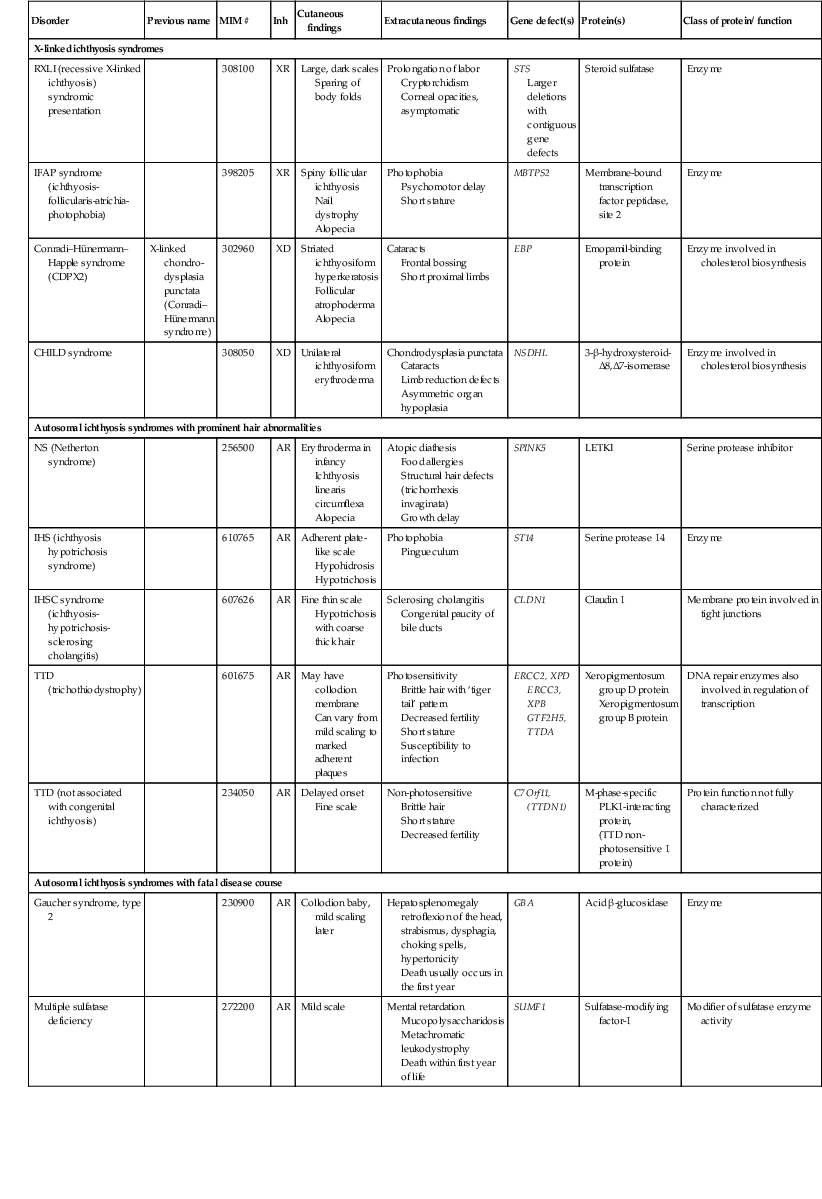
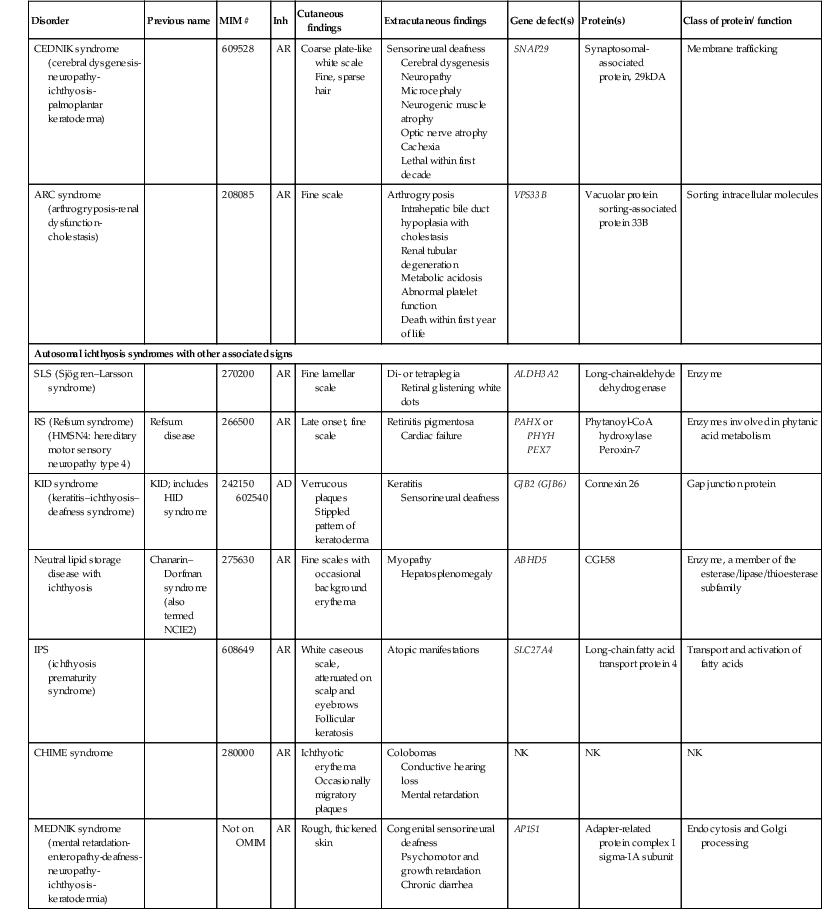
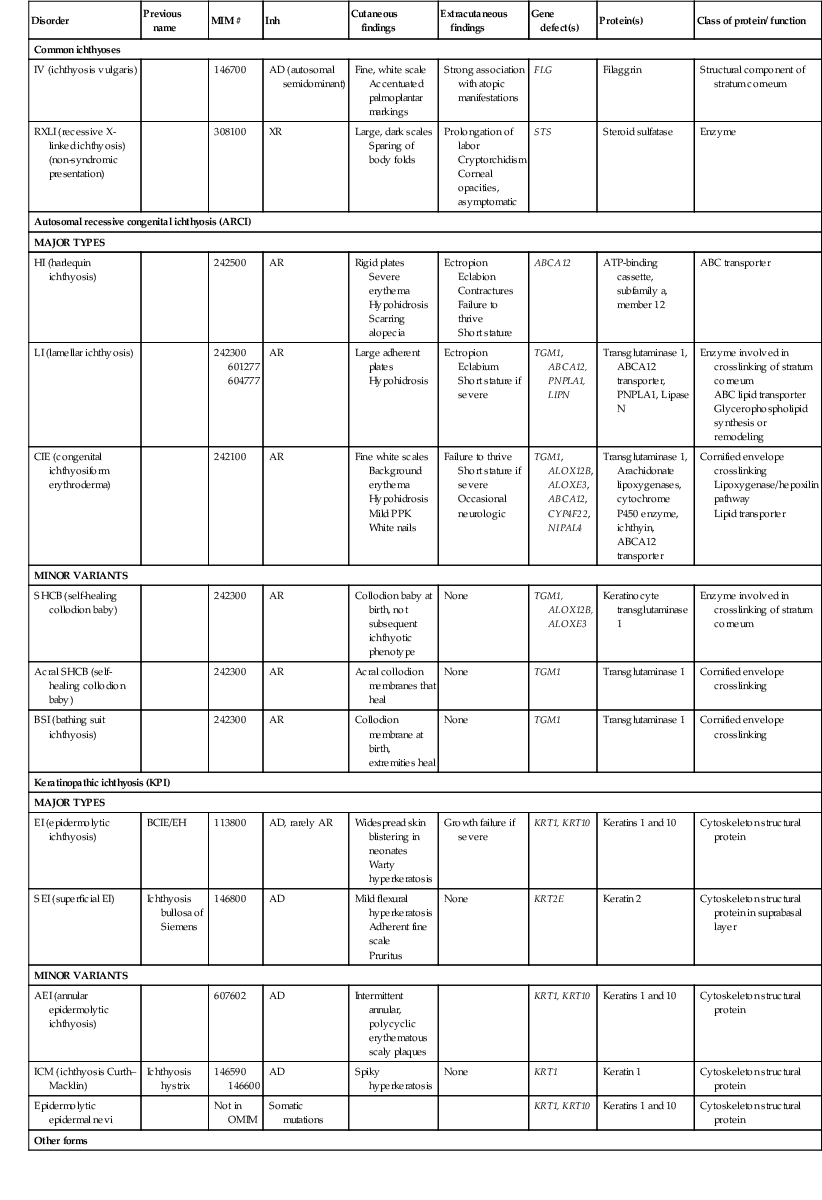
Disorders of Cornification (Ichthyosis)
Introduction
Collodion baby
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Ichthyosis vulgaris
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Recessive X-linked ichthyosis
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Inherited syndromic ichthyoses – X-linked
Ichthyosis follicularis, alopecia, and photophobia (IFAP syndrome)
Cutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
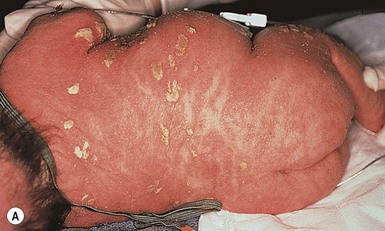
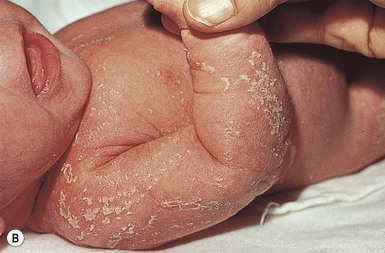
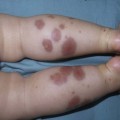
Conradi–hünermann–happle syndrome (X-linked chondrodysplasia punctata)
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
CHILD syndrome
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Inherited syndromic ichthyoses with prominent hair signs
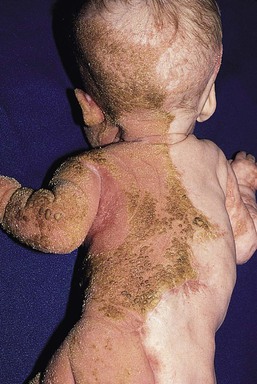
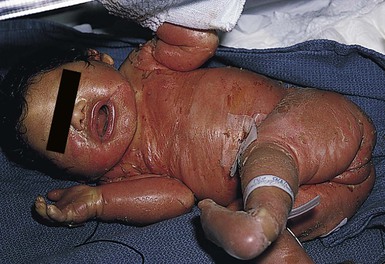
Netherton syndrome
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Ichthyosis hypotrichosis sclerosing cholangitis syndrome
Cutaneous features
Extracutaneous features
Etiology and pathogenesis
Differential diagnosis
Treatment and care
Trichothiodystrophy (IBIDS syndrome (Tay syndrome), PIBIDS syndrome, SIBIDS syndrome)
Cutaneous features
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of Cornification (Ichthyosis)
19