Autoinflammatory disorders are sterile inflammatory conditions characterized by episodes of early-onset fever, rash, and disease-specific patterns of organ inflammation. Gain-of-function mutations in innate danger-sensing pathways, including the inflammasomes and the nucleic acid sensing pathways, play critical roles in the pathogenesis of IL-1 and Type-I IFN-mediated disorders and point to an important role of excessive proinflammatory cytokine signaling, including interleukin (IL)-1b , Type-I interferons, IL-18, TNF and others in causing the organ specific immune dysregulation. The article discusses the concept of targeting proinflammatory cytokines and their signaling pathways with cytokine blocking treatments that have been life changing for some patients.
Key points
- •
Monogenic autoinflammatory disorders are hyperinflammatory conditions caused by single-gene mutations in innate immune regulatory pathways. They often present in infancy or early childhood; some can mimic infection or sepsis.
- •
Recognizing the clinical and histologic features is essential in initiating genetic testing, appropriate referral to make an early diagnosis, and to begin aggressive treatment to prevent organ damage and life-threatening consequences.
- •
Patients with interleukin (IL)-1–mediated disorders present with neutrophilic urticaria, pustulosis, and migratory rash, whereas interferon (IFN)-mediated disorders may present with panniculitis, livedo reticularis, or vasculitis.
- •
Anti–IL-1 therapy is the standard of care in treating the cryopyrinopathies (cryopyrin-associated periodic syndrome), and deficiency of IL-1 receptor antagonist and other IL-1–mediated conditions, including familial Mediterranean fever, hyperimmunoglobulinemia D syndrome, and tumor necrosis factor receptor–associated periodic syndrome.
- •
Discoveries of the genetic cause of novel autoinflammatory diseases suggest a role for type I IFN, IL-17, and IL-18; several have uncovered novel targets for therapeutic intervention.
| ADA2 | Adenosine deaminase 2 |
| AGS | Aicardi-Goutières syndrome |
| AID | Autoinflammatory disease |
| ANA | Antinuclear antibody |
| CAMPS | CARD14-mediated psoriasis |
| CANDLE | Chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperatures |
| CAPS | Cryopyrin-associated periodic syndrome |
| CINCA | Chronic infantile neurologic, cutaneous, and arthritis syndrome |
| CK | Creatinine kinase |
| CNS | Central nervous system |
| CRP | C-reactive protein |
| DADA2 | Deficiency of adenosine deaminase 2 (ADA2) |
| DIRA | Deficiency of interleukin-1 receptor antagonist |
| DITRA | Deficiency of interleukin-36 receptor antagonist |
| EO-IBD | Early onset of inflammatory bowel disease |
| ESR | Erythrocyte sedimentation rate |
| FCAS | Familial cold autoinflammatory syndrome |
| FDA | US Food and Drug Administration |
| FMF | Familial Mediterranean fever |
| GOF | Gain of function |
| HA20 | Haploinsufficiency of A20 |
| HIDS | Hyperimmunoglobulinemia D and periodic fever syndrome |
| IFN | Interferon |
| Ig | Immunoglobulin |
| IL | Interleukin |
| JMP | Joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy |
| LOF | Loss of function |
| MAS | Macrophage activation syndrome |
| MWS | Muckle-Wells syndrome |
| NLR | NOD-like receptor |
| NOMID | Neonatal-onset multisystem inflammatory disease |
| PAPA | Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome |
| PGA | Pediatric granulomatous arthritis |
| PLAID | PLACγ2-associated antibody deficiency and immune dysregulation |
| PRAAS | Proteasome-associated autoinflammatory syndrome |
| SAVI | Stimulator of interferon genes (STING)–associated vasculopathy with onset in infancy |
| SMS | Singleton-Merten syndrome |
| SPENCDI | Spondyloenchondrodysplasia with immune dysregulation |
| TNF | Tumor necrosis factor |
| TRAPS | TNF receptor–associated periodic syndrome |
Introduction
Autoinflammatory disorders are immune-dysregulatory conditions characterized by early onset, sterile systemic inflammatory episodes that include fever, rashes, joint pain, and features of disease-specific organ inflammation. The term autoinflammatory was proposed by Daniel Kastner in 1999 after the discovery of the genetic causes of the familial Mediterranean fever (FMF) and of the tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS).
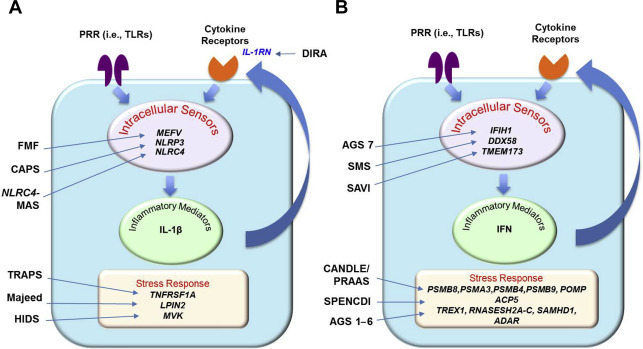
Over the last 15 years the discovery of the genetic causes for several autoinflammatory diseases revealed gain-of-function (GOF) mutations in intracellular innate immune sensors ( Fig. 1 ), including the interleukin (IL)-1 and IL-18 activating inflammasomes MEFV /pyrin, NLRP3 /cryopyrin, and nod-like receptor (NLR) family CARD domain-containing protein 4 ( NLRC4 ); NLRC4 also leads to high IL-18 serum levels. Mutations in the NOD-like receptor NOD2/caspase recruitment domain family, member 15 (CARD15) cause constitutive nuclear factor kappa B (NF-κB) activation; and more recently mutations in the viral RIG-I–like receptors, IFIH1/MDA-5 and DDX58, or the adaptor molecule TMEM173/STING encoding the stimulator of interferon genes (STING), are all linked to type I interferon (IFN) production and cause autoinflammatory phenotypes. Furthermore, loss-of-function (LOF) mutations in genes (ie, many of the enzymes) involved in protein or nucleic acid metabolism, cell transport, and other cellular homeostatic functions can lead to the accumulation of intracellular danger signals, which can trigger innate immune sensors and activate proinflammatory pathways (see Fig. 1 ). The increased and often constitutive release of the proinflammatory cytokines IL-1β, type I IFN, and TNF leads to autocrine cytokine amplification loops and these have become the targets for therapies. Although targeting IL-1 blockade therapeutically has become the treatment of choice for the IL-1–mediated diseases, targeting TNF is used empirically in some conditions and drugs targeting type I IFN or IL-18 are currently being used as compassionate use therapies.
Many monogenic autoinflammatory disorders present with characteristic skin rashes and cutaneous neutrophilic inflammation and patients are often referred to dermatologists for evaluation of the dermatologic findings and skin biopsies. It is therefore important for dermatologists to be familiar with these conditions because specific treatments can be life changing for many patients. This article presents groupings of autoinflammatory diseases based on dermatologic manifestations and fever patterns that allow a preliminary diagnosis based on systemic and dermatologic manifestations before genetic testing results become available ( Box 1 , Table 1 ).
- 1.
Nonspecific maculopapular rashes with recurrent episodic fever and abdominal pain (hereditary periodic fever syndrome)
- a.
Recurrent fever attacks of short duration (typically ≤7 days)
- i.
Familial Mediterranean fever
- ii.
Hyperimmunoglobulinemia D with periodic fever syndrome/mevalonate kinase deficiency
- i.
- b.
Recurrent fever attacks of longer duration (typically >7 days)
- i.
Tumor necrosis factor receptor–associated periodic syndrome
- i.
- a.
- 2.
Neutrophilic urticaria (CAPS)
- a.
Recurrent fever attacks of short duration (typically <24 hours)
- i.
CAPS/familial cold autoinflammatory syndrome
- ii.
CAPS/Muckle-Wells Syndrome
- i.
- b.
Continuous low-grade fever
- i.
CAPS/neonatal-onset multisystem inflammatory disease/CINCA
- ii.
IL-18–mediated AID and IL-1–mediated AID: NLRC4-related macrophage activation syndrome
- i.
- a.
- 3.
Pustular skin rashes and episodic fevers
- a.
IL-1–mediated pyogenic disorders with sterile osteomyelitis
- i.
Deficiency of IL-1 receptor antagonist
- ii.
Majeed syndrome
- i.
- b.
Partially IL-1–mediated pyogenic disorders
- i.
Pyogenic sterile arthritis, pyoderma gangrenosum, and acne syndrome
- ii.
Haploinsufficiency of A20 (monogenic form of Behçet disease)
- i.
- c.
Pyogenic disorders caused by non–IL-1 cytokine dysregulation
- i.
Deficiency of IL-36 receptor antagonist
- ii.
CARD14-mediated psoriasis (monogenic form of psoriasis)
- iii.
Early-onset inflammatory bowel disease
- i.
- a.
- 4.
Vasculopathy and panniculitis/lipoatrophy syndromes
- a.
Chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperature syndrome or proteasome-associated autoinflammatory syndrome
- a.
- 5.
Vasculopathy and/or vasculitis with livedo reticularis syndromes
- a.
Without significant CNS disease
- i.
STING-associated vasculopathy with onset in infancy
- i.
- b.
With severe CNS disease
- i.
Aicardi-Goutières syndrome
- ii.
Deficiency of adenosine deaminase 2
- iii.
Spondyloenchondrodysplasia with immune dysregulation
- i.
- a.
- 6.
Autoinflammatory disorders with granulomatous skin diseases
- a.
Without significant immunodeficiency
- i.
Blau syndrome (pediatric granulomatous arthritis, pediatric granulomatous arthritis)
- i.
- b.
With variable features of immunodeficiency and significant CNS disease
- i.
PLCγ2-associated antibody deficiency and immune dysregulation: cold-induced urticaria and/or granulomatous rash
- i.
- a.
- 7.
Other autoinflammatory syndromes
- a.
LACC1 -mediated monogenic Still disease
- a.
Abbreviations: AID, autoinflammatory disorder; CAPS, cryopyrin-associated periodic syndromes; CINCA, chronic infantile neurological cutaneous and articular syndrome; CNS, central nervous system.
| IL-1-mediated AIDs | IFN-mediated AIDs |
|---|---|
| Systemic | |
| CRP closely correlates with disease activity | CRP level only increased in severe disease |
| Granulocytosis with flares | Lymphopenia, leukopenia with flares |
| CNS | |
| Aseptic neutrophilic meningitis | Mild lymphocytic meningitis |
| Arachnoid adhesions (severe disease) | Basal ganglion calcifications, CNS vasculopathy, white matter disease |
| Other Organ Involvements | |
| Skin, vessels: neutrophilic dermatitis (urticarialike with mature neutrophilic infiltrates) | Panniculitis (immature neutrophils), lipoatrophy, vasculitis (chilblainlike lesions), microthrombotic disease |
| Lung/heart: serositis, pericarditis | Pulmonary fibrosis/interstitial lung disease, HTN, Pulmonary HTN |
| MSK: osteomyelitis, bony overgrowth, fasciitis | Myositis |
| ENT: hearing loss (inflammatory) | NA |
| Eyes: conjunctivitis, anterior uveitis | Glaucoma, episcleritis |
| Serology: 40% lupus anticoagulant positive | Some with increased autoantibodies |
Introduction
Autoinflammatory disorders are immune-dysregulatory conditions characterized by early onset, sterile systemic inflammatory episodes that include fever, rashes, joint pain, and features of disease-specific organ inflammation. The term autoinflammatory was proposed by Daniel Kastner in 1999 after the discovery of the genetic causes of the familial Mediterranean fever (FMF) and of the tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS).
Over the last 15 years the discovery of the genetic causes for several autoinflammatory diseases revealed gain-of-function (GOF) mutations in intracellular innate immune sensors ( Fig. 1 ), including the interleukin (IL)-1 and IL-18 activating inflammasomes MEFV /pyrin, NLRP3 /cryopyrin, and nod-like receptor (NLR) family CARD domain-containing protein 4 ( NLRC4 ); NLRC4 also leads to high IL-18 serum levels. Mutations in the NOD-like receptor NOD2/caspase recruitment domain family, member 15 (CARD15) cause constitutive nuclear factor kappa B (NF-κB) activation; and more recently mutations in the viral RIG-I–like receptors, IFIH1/MDA-5 and DDX58, or the adaptor molecule TMEM173/STING encoding the stimulator of interferon genes (STING), are all linked to type I interferon (IFN) production and cause autoinflammatory phenotypes. Furthermore, loss-of-function (LOF) mutations in genes (ie, many of the enzymes) involved in protein or nucleic acid metabolism, cell transport, and other cellular homeostatic functions can lead to the accumulation of intracellular danger signals, which can trigger innate immune sensors and activate proinflammatory pathways (see Fig. 1 ). The increased and often constitutive release of the proinflammatory cytokines IL-1β, type I IFN, and TNF leads to autocrine cytokine amplification loops and these have become the targets for therapies. Although targeting IL-1 blockade therapeutically has become the treatment of choice for the IL-1–mediated diseases, targeting TNF is used empirically in some conditions and drugs targeting type I IFN or IL-18 are currently being used as compassionate use therapies.
Many monogenic autoinflammatory disorders present with characteristic skin rashes and cutaneous neutrophilic inflammation and patients are often referred to dermatologists for evaluation of the dermatologic findings and skin biopsies. It is therefore important for dermatologists to be familiar with these conditions because specific treatments can be life changing for many patients. This article presents groupings of autoinflammatory diseases based on dermatologic manifestations and fever patterns that allow a preliminary diagnosis based on systemic and dermatologic manifestations before genetic testing results become available ( Box 1 , Table 1 ).
- 1.
Nonspecific maculopapular rashes with recurrent episodic fever and abdominal pain (hereditary periodic fever syndrome)
- a.
Recurrent fever attacks of short duration (typically ≤7 days)
- i.
Familial Mediterranean fever
- ii.
Hyperimmunoglobulinemia D with periodic fever syndrome/mevalonate kinase deficiency
- i.
- b.
Recurrent fever attacks of longer duration (typically >7 days)
- i.
Tumor necrosis factor receptor–associated periodic syndrome
- i.
- a.
- 2.
Neutrophilic urticaria (CAPS)
- a.
Recurrent fever attacks of short duration (typically <24 hours)
- i.
CAPS/familial cold autoinflammatory syndrome
- ii.
CAPS/Muckle-Wells Syndrome
- i.
- b.
Continuous low-grade fever
- i.
CAPS/neonatal-onset multisystem inflammatory disease/CINCA
- ii.
IL-18–mediated AID and IL-1–mediated AID: NLRC4-related macrophage activation syndrome
- i.
- a.
- 3.
Pustular skin rashes and episodic fevers
- a.
IL-1–mediated pyogenic disorders with sterile osteomyelitis
- i.
Deficiency of IL-1 receptor antagonist
- ii.
Majeed syndrome
- i.
- b.
Partially IL-1–mediated pyogenic disorders
- i.
Pyogenic sterile arthritis, pyoderma gangrenosum, and acne syndrome
- ii.
Haploinsufficiency of A20 (monogenic form of Behçet disease)
- i.
- c.
Pyogenic disorders caused by non–IL-1 cytokine dysregulation
- i.
Deficiency of IL-36 receptor antagonist
- ii.
CARD14-mediated psoriasis (monogenic form of psoriasis)
- iii.
Early-onset inflammatory bowel disease
- i.
- a.
- 4.
Vasculopathy and panniculitis/lipoatrophy syndromes
- a.
Chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperature syndrome or proteasome-associated autoinflammatory syndrome
- a.
- 5.
Vasculopathy and/or vasculitis with livedo reticularis syndromes
- a.
Without significant CNS disease
- i.
STING-associated vasculopathy with onset in infancy
- i.
- b.
With severe CNS disease
- i.
Aicardi-Goutières syndrome
- ii.
Deficiency of adenosine deaminase 2
- iii.
Spondyloenchondrodysplasia with immune dysregulation
- i.
- a.
- 6.
Autoinflammatory disorders with granulomatous skin diseases
- a.
Without significant immunodeficiency
- i.
Blau syndrome (pediatric granulomatous arthritis, pediatric granulomatous arthritis)
- i.
- b.
With variable features of immunodeficiency and significant CNS disease
- i.
PLCγ2-associated antibody deficiency and immune dysregulation: cold-induced urticaria and/or granulomatous rash
- i.
- a.
- 7.
Other autoinflammatory syndromes
- a.
LACC1 -mediated monogenic Still disease
- a.
Abbreviations: AID, autoinflammatory disorder; CAPS, cryopyrin-associated periodic syndromes; CINCA, chronic infantile neurological cutaneous and articular syndrome; CNS, central nervous system.
| IL-1-mediated AIDs | IFN-mediated AIDs |
|---|---|
| Systemic | |
| CRP closely correlates with disease activity | CRP level only increased in severe disease |
| Granulocytosis with flares | Lymphopenia, leukopenia with flares |
| CNS | |
| Aseptic neutrophilic meningitis | Mild lymphocytic meningitis |
| Arachnoid adhesions (severe disease) | Basal ganglion calcifications, CNS vasculopathy, white matter disease |
| Other Organ Involvements | |
| Skin, vessels: neutrophilic dermatitis (urticarialike with mature neutrophilic infiltrates) | Panniculitis (immature neutrophils), lipoatrophy, vasculitis (chilblainlike lesions), microthrombotic disease |
| Lung/heart: serositis, pericarditis | Pulmonary fibrosis/interstitial lung disease, HTN, Pulmonary HTN |
| MSK: osteomyelitis, bony overgrowth, fasciitis | Myositis |
| ENT: hearing loss (inflammatory) | NA |
| Eyes: conjunctivitis, anterior uveitis | Glaucoma, episcleritis |
| Serology: 40% lupus anticoagulant positive | Some with increased autoantibodies |
Interleukin-1–mediated autoinflammatory diseases
Nonspecific Maculopapular Rashes with Recurrent Episodic Fever and Abdominal Pain Hereditary Periodic Fever Syndromes
The hereditary fever syndromes are characterized by episodic and periodic high fever attacks that are accompanied by abdominal and/or chest pain followed by periods of remission or reduced inflammation. They include FMF, hyperimmunoglobulinemia D and periodic fever syndrome (HIDS), and TRAPS.
Familial Mediterranean fever
FMF is an early onset autosomal recessive disorder caused by GOF mutation in the MEFV gene, which encodes pyrin, which leads to unregulated release of proinflammatory cytokines, such as IL-1β, and uncontrolled inflammation. Autosomal dominant forms of the disease exist. The inflammatory attacks typically last for 3 to 7 days and recur every 4 to 6 weeks, and are accompanied by severe abdominal pain and to a lesser degree chest pain caused by sterile serositis. Debilitating myalgia with normal creatine kinase (CK) levels and sometimes synovitis is also seen. During the attacks, inflammatory markers, erythrocyte sedimentation rate (ESR), and C- reactive protein (CRP) level are highly increased and may normalize between attacks. Patients from Armenia, Turkey, and Arabian countries with a long-standing history of FMF are at high risk of developing AA amyloidosis leading to renal failure.
Cutaneous manifestations
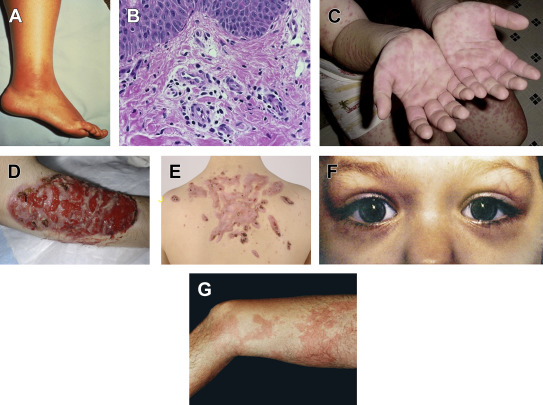
The prevalence of pathognomonic erysipelaslike erythema ( Fig. 2 A) varies widely among different populations (21% and 46% in Turks and Jews, but 3% and 8% in Arabs and Armenians ), and it occurs on the distal extremities. The lesions present as tender, warm, swollen, and erythematous plaques that are triggered by prolonged walking and subside within 24 to 48 hours. Other rarer eruptions include scattered nonspecific purpuric papules ; nodules have also been reported in children with FMF.
Related posts:
 Basic Science Insights into Clinical Puzzles
Basic Science Insights into Clinical Puzzles
 Establishing Tolerance to Commensal Skin Bacteria
Understanding Inherited Cylindromas
Establishing Tolerance to Commensal Skin Bacteria
Understanding Inherited Cylindromas
 Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
 Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


