Fig. 15.1
Effect of cytokines on T helper (Th) cell development. Schematic depicts a mature dendritic cell (APC) in the lymph node presenting antigen (black oval) in the context of an MHC class II molecule to a naïve Th cell. Damage- or pathogen-associated molecular patterns (DAMPs and PAMPs) promote maturation and cytokine production by the APC. Development of Th subsets (Th1, Th2, Th9, Th17), CD4+FoxP3+ regulatory T cells (Treg) and follicular helper cells (Tfh) is dictated by interaction with the APC and specific cytokines or combinations of cytokines including interleukins (IL) and transforming growth factor beta (TGF-β). These Th subsets in turn produce specific cytokines including interleukins, TGF-β and interferon gamma (IFNγ) (Adapted from figure courtesy of Mark H. Kaplan, Ph.D.)
Table 15.1
Role of selected cytokines in psoriasis or atopic dermatitis pathogenesis
Disease | Cytokine | Sources | Stimuli | Target | Effect | Tx |
|---|---|---|---|---|---|---|
Psoriasis | TNF | DC, M, K Th17, Th22 | TNF, IL-1, GM-CSF, DAMP, PAMP | K, DC, T | Chem; N recruit; K prolif; AMP; IL-1,-6,-20R,-23,-36, DC mig/mat, Th17/Th22 dev | Y |
IL-1 | K, M, T, DC, Ma | TNF, IL-1, -36, DAMP, PAMP | K, DC, M, N | Chem; TNF, AMP, IL-6, -17, -36; N act, recruit, survival; APC act | N | |
IL-6 | K, M, T, E | TNF, IL-1, -17, PAMP | T, B, M, N | Chem; IL-21; Th17 dev; N recruit | N | |
IL-12 | DC, M, N | PAMP | T, NK | IFNγ, IL-9 | Y | |
IL-17 | Th17, ILC3, Tc17, γδT, NKT | TNF, IL-1, -6, -23 | K, T, M | Chem; TNF, IL-1, -6, G(M)-CSF; AMP; recruit T, DC, N | CL | |
IL-20R | K, M | IL-1, -17, -22 TNF, wound, UVB, GM-CSF | K | ↓ IL-1, ↓ IL-17 | N | |
IL-22 | Th17, γδT, Th22, NK, ILC3 | IL-23 | K | K prolif/↓ K diff; AMP | CL | |
IL-23 | DC, M | PAMP, TNF, IL-12, CD40L | Th17, γδT, ILC3 | IL-17, -22; Th17 act | Y | |
IL-28 or 29 | DC, M, Th17 | PAMP | K, M, T | AVP | N | |
IL-36 | K | TNF, IL-1, -17 | K, M, DC | Chem; N, T, M recruit; IL-1, -6; APC act | CL | |
IFN-α/β | pDC | PAMP | mDC | IL-12, -23 | ||
AD | IL-4 | Th2, Ma, Ba, Eo | TCR act; FcεRI x-link; IL-33, TSLP, IL-25 | K, T, B, My | Th2 dev: BCR, MHCII, IgE in B; DC mat; Eo recruit; Ma prolif/survival; IL-10, -12; ↓ K diff/prolif → ↓barrier fxn | CL |
IL-5 | Th2, ILC2, Ma, NK, Eo | TCR act; FcεRI x-link: PAMP, IL-33, TSLP, IL-25 | Eo, B, Ma, Ba | Eo recruit, survival, act, B cell diff | N | |
IL-13 | Th2, ILC2, Ma, Ba, Eo, NKT | TCR act; FcεRI x-link: IL-33, TSLP, IL-25 | T, B, My | IgE, DC mat, Eo recruit, Ma act, FcεR and MHC II on B and M, | CL | |
IL-25 | Th2, Ma, NKT, K, DC, Eo, | TCR act; PAMPs | T, M, K, ILC2 | Th2 cytokines (IL-4, -5, -13); IgE, ↓ K diff/AMP | N | |
IL-31 | Th2, Ma | IL-4, -33, AMP | Neur, K, DC, M | Itch; ↓ K diff/prolif → ↓barrier fxn, Chem, | CL | |
IL-33 | K, Ma, E | IFNγ, IL-1, TNF, cell damage or death | ILC2, Th2, Ma, M, Eo, Ba, DC | Th2 cytokines (IL-4, -5, -13; -31); Th2 dev; Ma act, M2 dev; Eo recruit, | N | |
TSLP | K, DC | Epid injury, PAR2, PAMP | DC, Ma, Eo, Th2, ILC2, Treg | Itch, IL-4, -5, -13; Chem → Th2 recruit | CL |
JAK-STAT Signaling
Cells use cytokines to communicate with themselves and other cells via cytokine-specific receptors. Receptor engagement activates intracellular signaling pathways that dictate target cell responses. Regulation of target gene expression is an important effect of cytokine stimulation. Many cytokines, namely interleukins and interferons, use receptors that activate Janus kinases (JAK1, JAK2, JAK3 and Tyk2) to initiate signal transduction; Janus kinases in turn phosphorylate signal transducer and activator of transcription (STAT) proteins which include STAT1, STAT2, STAT3, STAT4, STAT5 and STAT6. Phosphorylated STAT proteins then homo- or heterodimerize and translocate to the nucleus where they regulate cytokine-dependent target gene expression (Fig. 15.2). Janus kinase inhibitors are currently in clinical trials for psoriasis and atopic dermatitis. Thus, understanding the concepts of how JAK and STAT proteins function will likely become more important to the practice of dermatology. The specific JAK and STAT proteins used by individual cytokines will be discussed in the coming sections.
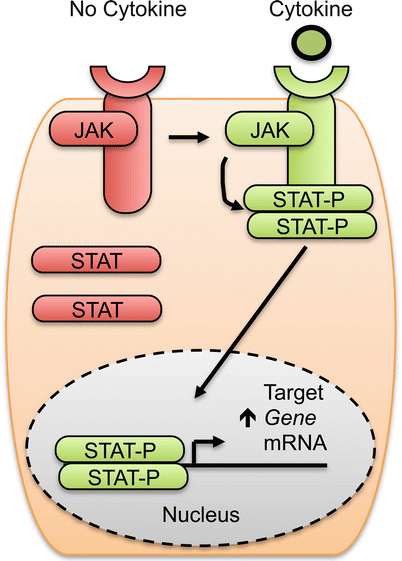
Fig. 15.2
Overview of Janus kinase (JAK) and signal transducer and activator (STAT) signaling pathway utilized by many cytokines. In the absence of cytokine, the cytokine receptor, JAKs and STATs are inactive (red shading). Cytokine (green circle) activates the pathway (green shading). Activated JAKs phosphorylate STAT proteins (designated STAT-P). STAT-P then dimerizes and translocates to the nucleus to regulate target gene expression
TNF Family
The tumor necrosis factor superfamily (TNFSF) originally consisted of TNF and lymphotoxin; at least 16 more members have been identified [1]. In addition to immunomodulation, some TNFSF members also regulate processes like organogenesis, epithelial cell development and bone remodeling [2]. Some of the other family members are linked to pathogenesis of rare diseases like X-linked hypohidrotic ectodermal dysplasia, Stevens-Johnson Syndrome/Toxic epidermal necrosis and hyper IgM syndrome, but the most important by far to the current understanding of cutaneous immunology, pathogenesis and therapy for dermatologic diseases is TNF [3–5].
The TNFSF is composed of cell surface-bound and/or soluble proteins, most of which are synthesized as heterotrimeric transmembrane proteins [6, 7]. The trimeric TNF homology domain of many TNFSF members can be liberated from the cell surface (i.e. solubilized) by specific proteases [6]. Surface bound and/or soluble forms of the trimeric TNF homology domains act as ligands for their cognate receptor(s) [7]. In humans, the TNF receptor superfamily (TNFRSF) includes at least 29 members [8]. Most are synthesized as transmembrane proteins; some undergo proteolysis to generate soluble receptors, while others are synthesized de novo as soluble receptors. The extracellular domain or soluble forms of these receptors bind TNFSF members, while the intracellular portions of the transmembrane forms mediate signal transduction. The TNFRSF are divided into activating and death receptors. Activating receptors trigger nuclear factor kappa B (NF-kB) and mitogen-activated protein kinase (MAPK) signaling, promoting cellular survival, proliferation and differentiation. Death receptors initiate apoptosis or necroptosis [8].
Tumor Necrosis Factor
Multiple cell types including macrophages, dendritic cells and keratinocytes produce TNF [9–11]. Production is regulated at transcriptional and post-transcriptional levels and is induced by cytokines (e.g. GM-CSF, IL-1 & TNF) and danger signals, the latter of which are subclassified as pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) such as lipopolysaccharide and the host protein HMGB1, respectively [12–15].
After synthesis, TNF is expressed on the cell surface as a homotrimer, which can activate TNF receptors directly or undergo site-specific cleavage by the proteases ADAM10 and 17 releasing a homotrimeric soluble form [13, 16, 17]. While the membrane-bound form is important for secondary lymphoid tissue development, defense against intracellular pathogens and limiting chronic inflammation and autoimmunity, the soluble form promotes acute and chronic inflammation [18, 19]. These differential effects are in part explained by the fact that the membrane-bound form of TNF can activate both TNF receptors (TNFR1 and TNFR2), while the soluble form can only engage TNFR2 [13, 20]. While TNFR1 is widely expressed on many cells types, TNFR2 expression is more restricted to leukocytes, endothelial cells, keratinocytes and several other cell types [21]. TNFR2 is an activating receptor, while TNFR1 is both a death and activating receptor.
Given the pleiotropic functions and numerous contexts in which TNF acts, it is beyond the scope of this chapter to discuss all that this molecule does in the skin. Instead its role in psoriasis will be presented as an example of how TNF regulates cutaneous inflammation (see also Chap. 21). Increased quantities of TNF mRNA and protein are detected in psoriatic lesions [22]. Potential sources of TNF in psoriatic lesions include myeloid cells (e.g. TNF/iNOS-producing dendritic cells), T helper cell subsets (Th17 and Th22) and keratinocytes [23, 24]. In response to TNF, keratinocytes can proliferate and produce chemokines, cytokines and antimicrobial peptides; chemokines produced by TNF-stimulated keratinocytes recruit myeloid dendritic cells, Th17 cells and neutrophils, promoting inflammation [23]. Conversely, TNF acts on immature dendritic cells in the skin to promote their maturation and migration out of the skin and into draining lymph nodes where they present antigen to naïve T cells [23]. In the lymph node, TNF can promote differentiation of Th17 and Th22 cells from naïve T cells [25]. The Th17 and Th22 subsets can then return to the skin where they produce effector cytokines including IL-17A, IL-17F and/or IL-22, that can stimulate keratinocytes, leading to epidermal hyperplasia, production of antimicrobial peptides, chemokines and other cytokines, further promoting inflammation and disease [23]. The pleiotropic functions of TNF in psoriasis pathogenesis demonstrate how this cytokine is a central mediator of disease, which helps explain why TNF inhibitors are so efficacious for treatment of psoriasis and other dermatologic conditions [26, 27].
Interleukins
The term Interleukin (meaning between leukocytes) was originally proposed in 1979 in an effort to distinguish between two biochemically distinct lymphocyte activating proteins, interleukin 1 (IL-1) and interleukin 2 (IL-2) [28]. Since then, the list of interleukins has grown, now ending at IL-38 [29]. Some interleukin designations are further subdivided (i.e. IL-1α and IL-1β or IL-36α, β, γ) as a result of identifying cytokines (encoded by different genes) that signal via the same receptor. Based on similarities in structure, function and/or receptor usage, interleukins can be subdivided into families. In this section, the defining characteristics of a given interleukin family will be presented followed by a more detailed discussion of the family members most relevant to understanding cutaneous immunity, inflammation and disease. The families discussed are, IL-1, common gamma chain (γc), beta chain, IL-6, IL-10, IL-12, and IL-17.
Interleukin 1 Family
The IL-1 family consists of structurally similar but functionally diverse cytokines with a number of critical pro- and anti-inflammatory actions that govern cutaneous inflammation, immunity and disease. In humans, there are at least 11 members of the IL-1 family, most of which are receptor agonists (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, IL-36γ and IL-37), with the remainder being receptor antagonists (IL-1Ra, IL-36Ra and IL-38) [29]. With the exception of IL-37, which can bind to IL-18α alone, IL-1 family members that act as receptor agonists bind heterodimeric receptors [29]. The receptor complexes for IL-1α, IL-1β, IL-33 and IL-36α, IL-36β and IL-36γ are composed of a ligand-selective subunit bound to IL-1 receptor accessory protein (IL-1RAcP), while the IL-18 receptor is composed of two unique subunits, IL-18Rα and IL-18Rβ. The IL-1 receptor family members consist of an extracellular immunoglobulin-like domain and an intracellular portion that includes a TIR domain, the latter of which is also present in toll-like receptors (TLR). The TIR domain recruits the adaptor protein MyD88 to initiate downstream signaling events leading to MAPK and NF-kB activation and subsequently, target gene expression [30]. Unique aspects of IL-1 family member synthesis, processing and release will be discussed in the coming sections.
Interleukin 1
The designation IL-1 refers to two structurally distinct proteins, IL-1α and IL-1β that are encoded by different genes but share common receptors (i.e. IL-1R1/IL-1RAcP or IL-1R2/IL-1RAcP receptor complexes) [30]. There is also an endogenous IL-1 receptor antagonist, IL-1Ra, a recombinant form of which has been used therapeutically to treat IL-1-mediated inflammatory diseases [31]. Similar to TNF, IL-1α and IL-1β are considered innate cytokines, playing a key role in promoting local and in some cases, systemic inflammation and immunity. In addition to contributing to homeostatic aspects of cutaneous immunity and inflammation, IL-1 is implicated in the pathogenesis of Still’s disease, pyoderma gangrenosum (PG), Behçet’s disease, hidradenitis suppurativa (HS) and a number of autoinflammatory diseases. Importantly, IL-1 blockade can be an effective therapy for these diseases [32–34]. Host defense against bacterial pathogens is also mediated by IL-1, as are various aspects of keratinocyte proliferation, differentiation and function (see also Chap. 16) [35–37].
Keratinocytes and other epithelial cell types constitutively produce IL-1α [38]. Unlike IL-1β and most other cytokines, IL-1α is synthesized as a precursor containing a nuclear localization sequence allowing IL-1α to traffic into the nucleus, bind chromatin and regulate transcription [29]. Cellular damage or necrosis promotes release of IL-1α into the local milieu thereby promoting local (neutrophil-mediated) inflammation; as such IL-1α is classified as an alarmin. In contrast, IL-1α remains tightly bound to chromatin during apoptosis, thus restricting inflammation [29].
The disparate effects of IL-1β, compared to IL-1α, are largely a function of differences in cellular processing and cellular sources rather than differences in signaling pathways used by these cytokines [29, 38]. Sources of IL-1β include monocytes, macrophages and dendritic cells and keratinocytes [30]. Like IL-1α, IL-1β is synthesized as a precursor. Unlike IL-1α, production and release of the active form of IL-1β is a highly regulated process mediated by an intracellular complex known as the inflammasome (Fig. 15.3). Production of IL-1β begins with transcription of the IL1B gene and synthesis of Pro-IL-1β. Multiple stimuli (referred to as signal 1) promote IL1B transcription (e.g. TNF, TLR ligands and IL-1) [29]. Unlike IL-1α, the IL-1β precursor (Pro-IL-1β) does not activate IL-1 receptors. Instead, Pro-IL-1β must first undergo proteolysis to generate mature IL-1β [39]. Proteolysis of Pro-IL-1β is performed by caspase-1, which itself must first undergo proteolytic cleavage by the inflammasome. Stimuli that trigger inflammasome activation (referred to here as signal 2) include DAMPs like ATP, pore forming bacterial toxins and others [36]. Upon activation, the inflammasome cleaves procaspase-1 to caspase-1; caspase-1 in turn cleaves Pro-IL-1β generating IL-1β, which is then secreted (Fig. 15.3).
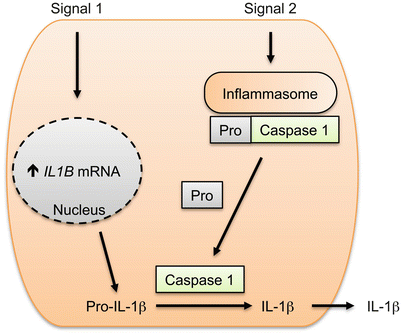
Fig. 15.3
Two-signal model for production of IL-1β in host cells. Signal 1 (e.g. cytokine) induces expression of IL1B mRNA, which is then translated in an inactive protein (Pro-IL-1β). Signal 2 (e.g. ATP) stimulates the same cell activating the inflammasome, which then generates Caspase 1 from its inactive precursor, Pro-Caspase 1. Caspase 1 then cleaves Pro-IL-1β, generating IL-1β that is then secreted from the cell
Gain of function mutations in genes encoding proteins in inflammasome underlie a number of autoinflammatory diseases with cutaneous manifestations including: pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome as well as the periodic fever syndromes, familial Mediterranean fever (FMF) and cold induced auto-inflammatory syndromes-1 (CIAS-1), like Muckle-Wells syndrome, familial cold urticaria and neonatal-onset multisystem inflammatory disease [40]. In patients with CIAS-1 spectrum disease, periodic fevers result from increased circulating levels of IL-1β acting as a pyrogen, while urticarial eruptions may be due to skin-resident mast cell-derived IL-1β stimulating increased permeability of cutaneous blood vessels [41, 42]. The critical role of IL-1β in autoinflammatory diseases is highlighted by their responsiveness to IL-1-blocking therapeutics [40].
Several key aspects of host defense against cutaneous infections with Staphylococcus aureus are dependent on IL-1 [36]. In this setting, cell wall components from S. aureus (e.g. lipotechoic acid and bacterial peptidoglycan) activate TLR2 on keratinocytes and macrophages stimulating IL-1 production [43]. Keratinocytes in turn produce IL-1α, and lesser amounts of IL-1β, while macrophages primarily produce IL-1β [36]. Activation of IL-1 receptors for IL-1α or IL-1β, then stimulates keratinocyte production of antimicrobial peptides, neutrophil-attracting chemokines and other cytokines, like IL-6. Antimicrobial peptides can directly combat the infection, while chemokines and IL-6 can recruit neutrophils into the skin; notably, IL-1 can also prolong neutrophil survival, further promoting neutrophil-mediated clearance of the infection [36, 44].
Interleukin 33
Interleukin 33 (IL-33) is a dual function protein that acts as a cytokine and a nuclear factor that may regulate NF-kB-dependent genes [45, 46]. Cellular damage or necrosis releases IL-33 promoting local inflammation; thus IL-33 is an alarmin like IL-1α [47]. Conversely, proteolytic cleavage inactivates IL-33 during apoptosis preventing IL-33-mediated inflammation [48]. The IL-33 receptor is expressed on Th2 cells, innate lymphoid type 2 cells (ILC2), macrophages, mast cells, eosinophils and other cells. Effects of receptor engagement are cell type specific explaining the pleiotropic functions of IL-33 [49]. For example, IL-33 promotes Th2 cell development and cytokine production, mast cell maturation and activation and polarization of macrophage development towards an M2 phenotype [50, 51]. Paradoxically, IL-33 can promote Th1-polarized inflammation via NK and CD8+ T cell stimulation [50]. Expression of IL-33 is increased in lesional skin keratinocytes in atopic dermatitis (AD), psoriasis and lichen planus [52, 53]. In AD, IL-33 concentrations are reportedly increased in the peripheral blood of patients with moderate to severe disease and are reduced following effective topical therapy with steroids or calcineurin inhibitors [52, 54]. These results suggest keratinocyte damage or death in AD lesional skin (possibly secondary to scratching the skin) release IL-33 into the circulation, the consequences of which are not defined in this context.
Interleukin 36
The IL-36 family includes receptor agonists IL-36α, IL-36β and IL-36γ, hereafter referred to as IL-36. The family also includes IL-36Ra, a receptor antagonist [55]. The IL-36 receptor is composed of IL-36R and IL-1RAcP and utilizes signal transduction pathways like that described above for other IL-1 family members. Loss of function mutations in the IL36RN gene (encodes IL-36Ra) cause some familial and sporadic cases of generalized pustular psoriasis and Acrodermatitis Continua of Hallopeau [56, 57]. Loss of IL-36Ra function is thought to lead to unopposed activation of IL-36 signaling.
The IL-36 receptor is expressed on keratinocytes, monocytes and some dendritic cells [55, 58, 59]. Keratinocytes can produce IL-36α, IL-36β, IL-36γ and IL-36Ra [55]. Transcripts encoding IL-36 family, in low abundance under non-inflammatory conditions, are increased under inflammatory conditions as seen in psoriatic lesions or keratinocytes stimulated with TNF, IL-17, or IL-1 [55]. In human keratinocytes, IL-36 stimulates production of chemokines known to attract T cells, neutrophils and macrophages [58]. In contrast, stimulation of monocytes and myeloid dendritic cells with IL-36 triggers IL-1β and IL-6 production and increased proportions of cells expressing the co-stimulatory molecule CD86 and MHC class II [58]. These observations suggest IL-36 family members are important mediators of inflammation in the skin. Consistent with results from genetic studies suggesting a role for IL-36 in psoriasis pathogenesis, IL-36 is reported to promote psoriasiform inflammation in mice [55–59]. Integrating our current understanding of psoriasis pathogenesis and IL-36 biology, the literature suggests the following role for IL-36 in disease: (1) innate cytokines like IL-1β and TNF as well as IL-17A can stimulate IL-36 production by keratinocytes; (2) IL-36 stimulates keratinocytes to produce multiple chemokines promoting migration of neutrophils, macrophages and T cells in and into the skin; (3) IL-36 stimulates some dendritic cell subsets promoting antigen presentation to T cells and production of innate cytokines like IL-1β and IL-6; (4) IL-1β, TNF, and IL-17A promote various aspects of inflammation in psoriatic lesions and further production of IL-36 by keratinocytes leading to a vicious cycle and persistent disease. Thus, IL-36 or its receptor may become viable therapeutic targets for psoriasis and potentially other dermatologic conditions. Future investigation is still needed to clarify the role of IL-36 in other aspects of normal or pathologic inflammation and immunity in the skin.
Common Gamma Chain Receptor Family
The common gamma chain (γc) receptor family includes IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 [60]. The γc receptor subunit that defines the family is a transmembrane protein that transduces signals in part through JAK3 [61]. Receptor signaling is initiated by heterodimerization or heterotrimerization with ligand-specific receptor subunits [62]. These receptors signal via JAK-STAT, PI3K-Akt and RAS/MAPK pathways promoting cell survival, proliferation and regulation of target gene expression [63]. The JAK-STAT family members activated by these cytokines is ligand-specific. Recently, a secreted form of γc generated by alternative splicing has been identified in mouse and human T cells and appears to act as a decoy receptor for some γc family cytokines [64].
Cytokines in the γc family regulate lymphoid cell development, survival and/or function (see also Chap. 8) [65]. Additional targets include mast cells, myeloid cells and other hematopoietic cells. Genetic deficiency of functional γc chains or JAK3, leads to severe combined immunodeficiency (SCID), highlighting the importance of this receptor family in host immunity and inflammation [66]. Cutaneous manifestations of these forms of SCID include eczematous dermatitis and infections with S. aureus, HPV or Candida spp. and graft versus host disease (GVHD) [67]. The diverse cutaneous and systemic manifestations seen in these forms of SCID reflect broad defects in IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 function.
In the coming sections, the roles of IL-2, IL-4 and IL-9 in cutaneous immunity and inflammation will be presented. The functions of IL-7, -15 and -21 in the skin are less well defined and will be discussed only briefly. Owing to overlap in cytokine receptor usage with some γc family members, IL-13 and TSLP will also be discussed in this section.
Interleukin 2
Interleukin 2 (IL-2) is the founding member of this family. Activated T cells produce IL-2, as do B cells and some dendritic cells [68–70]. There are two receptor complexes for IL-2, a heterodimer of the β and γc chains and a heterotrimer of the high affinity subunit IL-2Rα (i.e. CD25) bound to the β and γc chains [71]. While IL-2Rα increases binding affinity for IL-2, the β and γc chains regulate signal transduction. Shared usage of β and γc chains explains similarities in biologic effects of IL-2 and IL-15, while use of distinct high affinity receptor subunits (i.e. IL-2Rα vs. IL-15Rα) helps explain differences between these cytokines [72].
One or both forms of the IL-2 receptor are expressed on T cells, B cells, NK cells, monocytes and macrophages [71, 73, 74]. In addition to PI3K-Akt and RAS/MAPK signaling, receptor engagement activates JAK1 and JAK3 triggering phosphorylation of STAT1, STAT3 and STAT5. T cell survival and proliferation are important effects of IL-2 during antigen-specific immune responses, as in allergic contact dermatitis [75]. Based on the ability of IL-2 to promote T cell survival and proliferation, recombinant human IL-2 became one of the first FDA-approved immunotherapies for metastatic melanoma [76, 77]. In contrast, an IL-2-diphtheria toxin fusion protein is used to trigger death in cells expressing IL-2Rα; this agent is used to treat some forms of cutaneous T cell lymphoma (CTCL) [78]. Interleukin 2 also regulates peripheral tolerance through effects on regulatory T cells, promotion of Fas-mediated activation-induced cell death of Th cells, and by restraining CD8+ T cell responses [79, 80]. Some of the aforementioned immunostimulatory and immunosuppressive effects of IL-2 are lacking in IL-2Rα deficient patients, leading to features of autoimmunity and immunodeficiency as well as cutaneous manifestations including psoriasiform dermatitis and alopecia universalis [81].
Interleukin 4 and 13
Interleukin 4 (IL-4) is a canonical Th2 cytokine produced by Th2 cells, mast cells, basophils and eosinophils [82]. There are two functionally distinct heterodimeric IL-4 receptor complexes. The first (Type I receptor) is specific for IL-4 and is composed of IL-4Rα and γc; the second (Type II receptor) binds IL-4 or IL-13 and is composed of IL-4Rα and the IL-13Rα1 [83]. The Type I receptor is expressed on T and B cells, myeloid cells and fibroblasts and acts through JAK1 and JAK3 leading to phosphorylation of STAT3 and STAT6. The Type II receptor is also expressed by myeloid cells and fibroblasts and is selectively expressed on smooth muscle and epithelial cells, including keratinocytes and acts through JAK1, JAK2, JAK3 and TYK2 and activation of STAT3, STAT5 and STAT6 [82–84].
Effects of IL-4 are pleiotropic and cell-type dependent. Depending on the presence or absence of TGF-β, IL-4 can polarize naïve CD4+ Th cell differentiation towards Th2 or Th9 lineages; IL-4 can also promote T cell survival (Fig. 15.1) [85, 86]. In B cells, IL-4 promotes survival, B cell receptor and MHC class II expression, and IgE class switching [87–89]. Mast cell survival and proliferation are also promoted by IL-4 [90]. In keratinocytes, IL-4 impairs differentiation, which correlates with impaired epidermal barrier function in vivo [91–93]. Dendritic cell maturation and eosinophil chemotaxis are also stimulated by IL-4 [94, 95].
Interleukin 13 is another Th2 cytokine and is produced by Th2 cells, ILC2, mast cells, eosinophils, basophils and NKT cells [96, 97]. The Type II receptor (shared with IL-4), mediates many of the overlapping biologic effects of IL-4 and IL-13 including IgE class switching, dendritic cell maturation, mast cell activation and eosinophil recruitment [83]. A second receptor, IL-13Rα2, is IL-13-specific and is generally regarded as a decoy receptor; though some IL-13Rα2-specific functions have been demonstrated in mouse models of allergic lung inflammation [83, 98].
The aforementioned functions of IL-4 and IL-13 explain why they are key mediators of allergic inflammation in multiple tissues and are implicated in the pathogenesis of various aspects of atopy, including AD [99]. Both cytokines are detectable in acute and chronic AD lesions and mediate key aspects of AD pathogenesis, epidermal barrier dysfunction and Th2-polarized inflammation. A neutralizing antibody against IL-4Rα that prevents IL-4 and IL-13 signaling is in clinical trials for AD [100].
Interleukin 9
Interleukin 9 is produced by several Th subsets, most notably Th9 cells, and mast cells; of note Th9 cell development is dependent on IL-4 and TGF-β (Fig. 15.1) [86, 101]. Type I interferons, IL-1, IL-6, IL-10, IL-12, IL-21, IL-25 and LPS promote IL-9 production in relevant cells types [86, 102, 103]. The IL-9 receptor is a heterodimer of IL-9R and γc that signals via JAK1 and JAK3 and then, STAT1, STAT3 and STAT5 [86, 104]. This receptor can be expressed on mast cells, Th2 cells, Th17 cells, ILC2, B cells, smooth muscle cells and some epithelial cells [86, 105]. Stimulation with IL-9 promotes mast cell proliferation and production of proteases and a variety of cytokines including IL-1β, IL-5, IL-6, IL-13, TGF-β [106, 107]. This cytokine also promotes Th17 development, ILC2 survival and production of IgE and IgG by B cells [86, 105].
Recently, IL-9 was implicated in the pathogenesis of psoriasis and mouse models of melanoma [108–110]. With respect to psoriasis, Th9 cells were more abundant in psoriatic lesions, suggesting a role in disease [108, 111]. In mouse models of melanoma, IL-9 can exert anti-tumor effects; although the proposed mechanisms underlying these observations varied with one report demonstrating mast cell-dependent anti-tumor effects of IL-9 and another suggesting that IL-9 indirectly promotes effector CD8+ T cell recruitment into tumors [109, 110].
Thymic Stromal Lymphopoietin
Thymic stromal lymphopoietin (TSLP) is included in this section because its receptor is composed of IL-7Rα and a γc-like receptor-subunit, TSLPR. In contrast to IL-7, which regulates development, proliferation and/or survival of lymphoid cells, TSLP promotes allergic inflammation in skin and other epithelial surfaces [112–114]. Upon stimulation with TSLP, its receptor activates JAK1 and JAK2 and subsequently, STAT1, STAT3, STAT4, STAT5, and STAT6, which then regulate TSLP target genes [74].
Keratinocytes and dendritic cells can produce TSLP [115]. Production of TSLP by keratinocytes is increased in lesional skin of AD and Netherton syndrome, both of which are characterized by epidermal barrier dysfunction and Th2-polarized inflammation [115, 116] (see also Chap. 22). Interestingly, experimentally induced epidermal barrier disruption and/or injury trigger TSLP production by keratinocytes in normal human skin [117]. In Netherton syndrome and AD, excessive serine protease activity can also stimulate the protease activated receptor 2 and consequently, TSLP production by keratinocytes. Activation of TLR2 signaling in keratinocytes by membrane fragments of S. aureus, a common skin pathogen that also colonizes the skin of patients with AD, can stimulate TSLP production [118]. Inflammatory cytokines can also promote TSLP production by keratinocytes and dendritic cells [114]. Thus, there are multiple potential stimuli to explain the increased production of TSLP by keratinocytes in lesional skin of patients with AD and Netherton syndrome, the downstream consequences of which will be described below.
Dendritic cells, Th2 cells, regulatory T cells, ILC2, mast cells, eosinophils, B cells and NKT cells express the TSLP receptor [114, 119]. Stimulation of dendritic cells with TSLP polarizes naïve T cell differentiation towards Th2 cells and promotes production of the chemokines CCL17 and CCL22, which can act as chemoattractants for Th2 cells. Moreover, stimulation of Th2, ILC2 and mast cells promotes Th2 cytokine production. Several mouse models of AD-like inflammation demonstrate that keratinocyte-derived TSLP production is sufficient to drive allergic inflammation, even in the absence of T cells [120]. Recently, TSLP was shown to directly stimulate sensory neurons in the skin leading to pruritus (itch) in mice injected with recombinant TSLP [121]. These findings indicate TSLP can directly promote allergic inflammation (via actions on leukocytes) and itch (via actions on neurons); both properties of TSLP are likely to be germane to AD pathogenesis. A phase I clinical trial with a humanized TSLP neutralizing antibody has been completed with healthy patients and patients with AD; however the results of these studies are not available at ClinicalTrials.gov.
Beta Chain Family: Interleukin 5
This family includes IL-3, IL-5 and granulocyte monocyte colony stimulating factor (GM-CSF) and is defined by use of the common β chain receptor (different from IL-2 receptor β chain) [122]. The common β chain receptor is widely expressed in hematopoietic cells, while expression of ligand-specific subunits is more restricted [123]. Functional receptors are heterodimers of the ligand-specific subunit and the common β chain [122]. Sources of IL-5 include Th2 cells, ILC2, mast cells, NK cells, CD8+ T cells, eosinophils and endothelial cells [96, 123]. The IL-5 receptor is composed of IL-5Rα and common β chain subunits; receptor engagement activates JAK1, JAK2, STAT1, STAT3 and STAT5 as well as RAS-MAPK and PI3K signaling [123]. As in other tissues, IL-5 promotes infiltration, survival and effector functions of eosinophils in the skin [123]. Like other Th2 cytokines, IL-5 is implicated in AD pathogenesis. However, treatment of moderate to severe disease with two weekly doses of an IL-5 neutralizing antibody failed to achieve the desired clinical endpoint suggesting IL-5 neutralization may not be sufficient to treat AD [124]. Interestingly, peripheral eosinophilia rapidly improved in patients treated with this antibody. While tissue eosinophilia was not assessed and the treatment course was short, these findings suggest eosinophils may not be required for persistence of AD lesions [124].
Interleukin 6 Family
This family is defined by shared receptor subunit usage amongst all or some of its members. Interleukins in this family include IL-6, IL-11, IL-30 and IL-31. Members that are not interleukins include oncostatin M, leukemia inhibitory factor and others [125, 126]. Most family members use a common receptor subunit named glycoprotein 130 (gp130), while other members (e.g. IL-31) are included in this family due to structural similarities with IL-6 or shared usage of receptor subunits with other family members and/or usage of gp130-receptor subunits [125, 126]. This discussion will focus on IL-6 and IL-31, both of which are implicated in important aspects of cutaneous immunity and inflammation in health and/or disease [125, 126].
Interleukin 6
A variety of cells produce IL-6 including lymphocytes, monocytes, macrophages, fibroblasts, keratinocytes, endothelial cells and others [125]. Stimuli for IL-6 production include PAMPs (e.g. LPS), cytokines (e.g. IL-1, TNF, IL-17) and others. The functional repertoire of IL-6 is extensive with effects on the acute phase response, thermal regulation, angiogenesis, pathologic bone resorption, lipid metabolism and many other physiologic and pathophysiologic processes. However, this section will focus on functions of IL-6 relevant to dermatologic diseases [125].
The IL-6 receptor is composed of gp130 and IL-6R. While gp130 is widely expressed, IL-6R is primarily restricted to lymphocytes, monocytes and neutrophils [125]. Receptor engagement activates JAK3, STAT3, MAPK and PI3K/Akt pathways [127]. A soluble form of the IL-6R subunit (similar to IL-15Rα) can also initiate signaling but the details of this are beyond the scope of this discussion [128]. Effects of IL-6 receptor activation are cell type specific. In B cells, IL-6 stimulates immunoglobulin production and plasma cell differentiation; the role of IL-6 in plasma cell differentiation may be an indirect effect mediated by IL-21 produced by IL-6 stimulated T cells [129]. Other effects of IL-6 on the T cell compartment are promotion of Th17 and follicular helper T cell (Tfh) cell development and suppression of regulatory T cell (Treg) development and function (Fig. 15.1) [130, 131]. As described below, IL-6 also promotes neutrophil accumulation in the skin [125].
While IL-6 can contribute to many normal (e.g. promotion of sterile inflammation after wounding) and pathologic processes in the skin, this section will focus on the roles of IL-6 in cutaneous bacterial infections and psoriasis. As noted earlier, IL-1 is a central mediator of host defense following cutaneous infections with S. aureus. One of the downstream mediators of this host defense mechanism is IL-6, which is produced by keratinocytes and myeloid cells in response to IL-1 [36]. Like IL-1, IL-6 stimulates production of chemokines that attract neutrophils; moreover, IL-6 stimulates endothelial cells to induce expression of adhesion molecules needed for neutrophil migration into the skin from the blood. The Th17 cell subset is thought to be an important source of IL-17A and IL-17F during cutaneous infections and in psoriatic lesions. Differentiation of Th17 cells from naïve T cells is promoted by IL-6, suggesting another, albeit indirect, mechanism whereby IL-6 can contribute to host defense against cutaneous bacterial infections and psoriasis pathogenesis (Fig. 15.1) [36]. For completeness sake, it should be noted that IL-17 producing cell types other than Th17 cells are also implicated in pathogenesis of cutaneous infections and psoriasis pathogenesis and that these populations do not appear to be IL-6 dependent [36, 132].
Interleukin 31
Like other Th2 cytokines, IL-31 is an important mediator of allergic inflammation and has been implicated in AD pathogenesis (see also Chap. 22) [126]. The Th2 cell subset produces IL-31 in response to IL-4 or IL-33, and mast cells can produce IL-31 upon stimulation with the antimicrobial peptides LL-37 and β-defensins [133, 134]. The IL-31 receptor is expressed on neurons, keratinocytes and some dendritic cell subsets, monocytes and macrophages [126, 135–137]. In contrast to most IL-6 family members, the IL-31 receptor does not utilize gp130; instead it is composed of a gp130-like subunit, IL-31Rα, and the oncostatin M receptor [138]. Receptor engagement activates JAK1, JAK2, STAT1, STAT3, STAT5, PI3K/Akt and MAPK signaling [138]. Like TSLP, IL-31 can directly stimulate neurons and provoke scratching of the skin in mice, suggesting IL-31 can directly induce pruritus (see also Chap. 12) [126, 139]. In vitro studies also suggest IL-31 impairs keratinocyte proliferation and differentiation, which may impair epidermal barrier function [135]. Finally, stimulation of CD1c+ dendritic cells with IL-31 promotes production of inflammatory cytokines and chemokines [137].
Pruritus is a characteristic feature of AD; IL-31 and TSLP are proposed to be important triggers for pruritus in this disease (i.e. they are pruritogens). In support of this, IL-31 expression is increased in lesional and nonlesional skin of patients with AD and in lesions of prurigo nodularis [126]. Serum levels of IL-31 are also elevated in AD and correlate with disease severity [140]. Based on the functions mentioned above, it is also possible that IL-31 contributes to pathogenesis via impairment of epidermal barrier function and promotion of cytokine and chemokine production. According to ClinicalTrials.gov, patients are currently being recruited for a phase I clinical trial of an IL-31 blocking antibody in healthy subjects and patients with AD.
Interleukin 10 Family
The IL-10 family includes IL-10, the IL-20 subfamily, which includes IL-19, IL-20, IL-22, IL-24, IL-26 and the type III interferon subfamily consisting of IL-28A, IL-28B and IL-29 [141]. This cytokine family is defined by common usage of type 2 cytokine receptors and structural similarities, the details of which will not be discussed in this chapter [141]. Despite structural similarities between these cytokines and similarities in receptor usage, the functions of many IL-10 family members are quite varied. This section will discuss IL-10 and IL-22 individually and then the IL-20 receptor cytokines, IL-19, IL-20 and IL-24. Though the details of type III interferons biology will not be reviewed here, these cytokines have recently been implicated in psoriasis pathogenesis and host defense against cutaneous viral infections; the reader is referred to the cited references for further information on these cytokines [141–147].
Interleukin 10
Interleukin 10 is one of the prototypic anti-inflammatory cytokines [141]. Multiple cells can produce IL-10 including lymphocytes (T, B & NK cells), macrophages, dendritic cells, mast cells, eosinophils, neutrophils and keratinocytes [141, 148]. The inductive stimuli regulating IL-10 production vary by cell type. In Th2 cells, IL-4 and IL-27 can stimulate IL-10 production; IL-6 and IL-27 can also stimulate IL-10 production by Th1 cells and Th17 cells [149, 150]. In addition to Th cells, several regulatory T cell subsets can produce IL-10; for instance, CD4+FoxP3+ regulatory T (Treg) cells produce IL-10 in response to IL-2 and IL-4 [151, 152]. Regulatory B cells also produce IL-10 [153]. In macrophages and dendritic cells, TLR (e.g. TLR2, TLR3, TLR4 and TLR9) engagement can also stimulate IL-10 production [154, 155].
The IL-10 receptor is primarily expressed on lymphoid and myeloid cell lineage cells and is composed of IL-10R1 and IL-10R2 [141, 156]. Receptor engagement by an IL-10 homodimer activates JAK1, TYK2, STAT1, STAT3 and STAT5 [157]. Two anti-inflammatory mechanisms by which IL-10 acts include the inhibition of various aspects of antigen presentation by macrophages and dendritic cells and attenuation of cytokine production by T cells [157]. Interestingly, multiple viruses, including Epstein Barr virus and cytomegalovirus produce IL-10 homologs, which are thought to attenuate antiviral immune responses [158]. With respect to dermatologic disease, the anti-inflammatory effects of IL-10 are likely to play a role in regulating many aspects of cutaneous inflammation and immunity. The list of dermatologic diseases in which IL-10 is implicated as a mediator of pathogenesis or a potential therapy is long including multiple infectious, inflammatory, autoimmune and neoplastic conditions [159]. Notably, clinical trials with recombinant IL-10 for psoriasis or wound healing did not show significant benefit [160].
Interleukin 22
Cellular sources of IL-22 include T cells (e.g. Th17, Th22, γδT cells), NK cells, and a subset of ILC3 [161–165]. Development of IL-22 producing cells and/or production of IL-22 are regulated by cytokines in a cell-type dependent manner. Development of Th17 cells, which produce IL-17 and IL-22, is driven by IL-6, IL-21 and TGF-β; whereas, development of Th22 cells, which produce IL-22 but not IL-17, is stimulated by IL-6 and TNF (Fig. 15.1) [25, 166–169]. Stimulation with IL-23 can induce IL-22 production by some T cell subsets and ILC3 [170–172]. While TGF-β promotes Th17 development, it inhibits IL-22 production by Th17 and Th22 cells [25, 173].
The IL-22 receptor, composed of IL-10R2 and IL-22R, is expressed on keratinocytes and other epithelial cells [141, 156, 174]. Receptor activation signals through JAK1, TYK2, STAT1, STAT3 and STAT5 and MAPK and Akt pathways [141]. In keratinocytes, IL-22 impairs differentiation and promotes proliferation (and thus epidermal hyperplasia) and increases expression of β-defensin 2, β-defensin 3, S100A8 and S100A9, all of which are molecular, cellular, or histologic features of psoriatic lesions [173, 175–177].
Psoriasis, AD and other diseases are mediated, in part, by IL-22 (see also Chap. 21) [23, 178]. In psoriasis and AD, increased levels of IL-22 and IL-22 producing cell subsets are described in lesional skin, and lesion resolution is associated with reduced IL-22 expression [23, 162, 178–186]. In both diseases, IL-22 is thought to act on keratinocytes leading to acanthosis and increased expression of S100A8, S100A9 and other proteins. While IL-22 can promote production of β-defensin 2 and β-defensin 3, expression is higher in psoriatic lesions than in AD; this may be due to Th2 cytokines attenuating expression of these β-defensins in AD [177, 187–189]. According to ClinicalTrials.gov AD patients are being enrolled for a clinical trial using an antibody to IL-22.
Interleukins 19, 20 & 24 (IL-20 Receptor Cytokines)
Interleukins 19, 20 and 24, hereafter referred to as IL-20 receptor cytokines, are discussed together due to overlapping receptor usage and biologic effects. Keratinocytes and other epithelial cells, monocytes, some T cells and melanocytes can produce one or more IL-20 receptor cytokines; myeloid dendritic cells expressing IL-20 are also present in psoriatic lesions [190–196]. In keratinocytes, IL-1β, IL-17A, IL-22, UVB light and wounding promote IL-20 receptor cytokine production [191, 197–199]. In monocytes, LPS, GM-CSF and TNF stimulate production [141, 196, 200, 201]. Both receptors for these cytokines are expressed by keratinocytes [176, 191]. Receptor engagement activates STAT3 driving keratinocyte proliferation and epidermal hyperplasia [176, 191]. Other effects of one or more of these cytokines on keratinocytes include increased expression of S100A7, as well as TNFA and MRP14 transcripts [176, 202].
Similar to IL-22, IL-20 receptor cytokines are implicated in psoriasis pathogenesis with expression of all three cytokines being increased in lesional skin [191, 196]. These cytokines are thought to contribute to epidermal hyperplasia in psoriatic lesions [176]. Consistent with this idea, local or systemic therapy for psoriasis reduces both epidermal hyperplasia and expression of these cytokines [203, 204]. It is uncertain whether targeting IL-20 receptor cytokines is a viable therapeutic option for psoriasis. According to ClinicalTrials.gov, one clinical trial with an anti-IL-20 blocking antibody was discontinued due to apparent lack of response; however, it is possible that IL-19 or IL-24 activities are sufficiently redundant to compensate despite IL-20 blockade. Paradoxically, IL-20 receptor cytokines were recently shown to exacerbate cutaneous infections with S. aureus in mice [205]. This is attributed to IL-20 receptor-mediated attenuation of IL-1β production, thereby decreasing the frequency of IL-17A producing T cells, which are important for host defense against infections with S. aureus [36, 205, 206]. This suggests the IL-20 receptor may be a viable therapeutic target for management of cutaneous infections with S. aureus.
Interleukin 12 Family
This family includes IL-12, IL-23, IL-27 and IL-35, all of which are heterodimers generated from the following subunits: p19, p28 (i.e. Ebi3) p35 (i.e. IL-12p35) and p40 (i.e. IL-12p40) [207]. Interleukins 12 and 23 are important pro-inflammatory cytokines in psoriasis and other diseases, and neutralizing antibodies against these cytokines are used to treat several of these diseases including psoriasis [208–211]. Understanding the roles of IL-12 and IL-23 in psoriasis and host defense against mycobacterial infections helps the clinician use these biologics effectively and safely. The other members of this family, IL-27 and IL-35, are primarily anti-inflammatory cytokines, but their roles in cutaneous immunology are less well defined and will not be discussed further in this chapter [207].
Interleukin 12
Interleukin 12 (IL-12p70) is composed of p40 and p35 and expressed by dendritic cells, monocytes, macrophages, B cells and neutrophils [212]. Stimulation of macrophages and dendritic cells with a variety of PAMPs causes IL-12 production [213, 214]. Cytokines (i.e. IFNγ, IL-4 and IL-13) and physical contact with T cells can also promote IL-12 production [215–217]. The IL-12 receptor, composed of IL-12Rβ1 and IL-12Rβ2, is expressed on T and NK cells [207]. Receptor engagement activates JAK2, Tyk2 and STAT4, triggering IFNγ production by T and NK cells [207, 218]. Stimulation of dendritic cells and monocytes with IFNγ can in turn stimulate additional IL-12 production resulting in a positive feedback loop [216]. Interleukin 12 also orchestrates differentiation of naïve T helper cells into Th1 cells, which are key producers of IFNγ (Fig. 15.1) [219, 220]. The ability of microbial products to trigger IL-12 production by dendritic cells and IL-12 to in turn dictate naïve T cell differentiation to Th1 cells is an example how cytokines link innate and adaptive immune responses.
Multiple dermatologic conditions are mediated by IL-12, some of which have been successfully treated with a p40 neutralizing antibody, which blocks both IL-12 and IL-23 [221–227]. As noted above, multiple clinical trials have established the efficacy of this antibody for psoriasis; there are case reports of therapeutic benefit in AD, HS and Behçet’s disease as well [228–230]. Host defense against mycobacterial infections is also critically dependent on IL-12-driven production of IFNγ, as discussed later in this chapter.
Interleukin 23
Interleukin 23 is a heterodimer of p19 and p40, the latter of which is shared with IL-12 [231]. Dendritic cells and macrophages produce IL-23 following activation of various pattern recognition receptors (e.g. TLR4), cytokine stimulation (e.g. TNF and IL-12) and contact-dependent interactions with T cells [212, 231, 232]. The IL-23 receptor is expressed on Th17 cells, some γδ T cells and ILC3 and is composed of IL-23R, and IL-12Rβ1; receptor engagement signals through JAK2, Tyk2, STAT3 and STAT4 [166, 231, 233–235]. Activation of STAT3 in naïve T cells with specific cytokines (e.g. IL-6) drives Th17 differentiation and IL-23 receptor expression; moreover, IL-23 stimulation further promotes IL-23R expression in these cells [166, 236]. In contrast, the IL-23 receptor is stably expressed on a subset of γδ T cells and is expressed on a subset of ILC3 [235, 237]. In Th17 cells and IL-23R+ γδ T cells, IL-23 can promote expansion of these subsets and IL-17 production [166, 235, 236]. Moreover, IL-23 stimulates IL-22 production by IL-23R+ ILC3 [237].
Interleukin 23 is implicated in the pathogenesis of psoriasis and many other dermatologic diseases [223, 224, 226, 227, 238–242]. Expression of both IL-23 subunits is increased in psoriatic lesions [238]. Moreover, several IL-23 responsive subsets of IL-17 producing cells are more abundant in psoriatic lesions; IL-17 and IL-22 expression are also increased in lesional skin [235, 237]. As noted, neutralization of p40 and thus IL-12 and IL-23 is an effective therapy for psoriasis. While, both cytokines likely contribute to disease, IL-23 is probably more important as demonstrated by the therapeutic efficacy of an IL-23 neutralizing antibody for psoriasis [243, 244]. In psoriasis, IL-23 is thought to drive IL-17 production; the therapeutic efficacy of IL-17 and IL-17 receptor blocking antibodies in clinical trials (discussed below) for psoriasis further supports the conclusion that IL-23 plays a more dominant role in psoriasis pathogenesis than does IL-12 (see also Chap. 43).
Interleukin 17 Family
The 6 members of this family are designated IL-17A, IL-17B, IL-17C, IL-17D, IL-17E and IL-17F; IL-17E is also known as IL-25 [245]. Emerging data suggest IL-25 can promote allergic inflammation in the skin; the reader is referred to the cited references for further information [120, 245–252]. The roles of IL-17B thru D in cutaneous immunology are not well established and will not be discussed further in this chapter. Family members IL-17A and IL-17F are important regulators of cutaneous immunity, inflammation and disease and will be discussed jointly due to shared receptor usage and biologic functions.
Interleukins 17A & 17F
Interleukin 17A, also known as IL-17, activates the same receptor as IL-17F, explaining the similar biologic functions of these cytokines. These cytokines can exist as homodimers or heterodimers of IL-17A and F [245]. Cells that produce IL-17A and/or F include several T cells subsets (i.e. Th17, Tc17 and γδTcells), NKT cells and a subset of ILC3 [165, 253–258] Investigations of IL-17 biology identified Th17 cells, a novel developmentally and functionally distinct population of T helper cells differentiated from naïve T cells stimulated with IL-1β, IL-6, IL-21, IL-23 and/or TGF-β; Th17 cell can produce IL-17A and F (Fig. 15.1) [253, 259]. Activation of the TCR and/or stimulation with IL-1 or IL23 can stimulate IL-17 production by Th17 cells, NKT cells and γδTcells [257, 260–262].
The receptor for IL-17A and F, referred to here as the IL-17 receptor, is a heterodimer of IL-17RA and IL-17RC [263]. T cells, B cells, macrophages, endothelial cells, fibroblasts and epithelial cells, including keratinocytes, express the IL-17 receptor [245]. Receptor engagement in keratinocytes recruits the adaptor protein Act1 and TRAF6 stimulating TAK1-mediated activation of NF-kB and C/EBP and MAPK-mediated activation of AP-1, culminating in transcriptional changes in IL-17 target genes [264–266]. Target genes induced by IL-17 receptor activation include those encoding pro-inflammatory cytokines (TNF, IL-1, IL-6, G-CSF, GM-CSF), chemokines (CXCL1, 3, 5, 6 and 8; CCL2, 7 and 20), antimicrobial peptides/proteins and matrix metalloproteinases [62, 245, 267–269]. Many chemokines produced by IL-17-stimulated keratinocytes recruit neutrophils into the skin, while keratinocyte-derived CCL20 can recruit dendritic cells and CCR6+ T cell subsets, including Th17 cells [268].
While IL-17A and F are implicated in the pathogenesis of other dermatologic conditions, this section will focus on the role of these cytokines in psoriasis and cutaneous infections [23, 36, 224, 270–273]. In psoriasis, IL-17 is thought to stimulate keratinocyte-derived chemokine and growth factor production (i.e. G-CSF & GM-CSF) leading to recruitment of neutrophils, T cells (e.g. Th17 cells) and dendritic cells into lesional skin [23]. Cellular sources of IL-17 in psoriasis include Th17 cells, γδT cells, NKT cells and ILC3 [23, 274]. Data from clinical trials suggest neutralizing antibodies to IL-17A and the IL-17 receptor (anti-IL-17RA) will be effective therapies for psoriasis (see also Chap. 43) [210, 275–277].
With respect to cutaneous infections, IL-17A and/or F play important roles in host defense against S. aureus and Candida spp. [36, 273]. As noted, neutrophil influx into the skin, a critical aspect of host defense against S. aureus infections, is regulated by chemokines and growth factors produced by IL-17 stimulated keratinocytes [36]. Host defense against cutaneous Candida spp. infections is also regulated by IL-17A and IL-17F as defective IL-17 signaling can cause chronic mucocutaneous candidiasis (CMC) [273]. In CMC, mutations in IL-17RA and IL-17F have been identified in some patients, while others have mutations that impair development of IL-17 producing T cells or mutations that lead to development of IL-17 neutralizing antibodies [273].
Interferon Family
Interferons were originally described over 50 years ago as soluble cell/tissue-derived factors that interfered with the ability of viruses to infect other cells or tissues [278]. Since that time, over a dozen interferons have been identified in humans. This section will focus on the two canonical classes of interferons, types I and II, which are distinguished by receptor usage and partially divergent downstream signaling pathways. There are over a dozen type I interferons in humans; all use the same receptor. In contrast, there is only one type II interferon, IFNγ, which uses a unique receptor. Interferons are important for various aspects of host defense against pathogens and are involved in pathogenesis of multiple inflammatory diseases. As such, the following sections will provide more detail on the biology of type I and type II interferons and the dermatologic diseases associated with each type.
Type I Interferons
Within this family are multiple interferon alpha (IFN-α) proteins and IFN-β, as well as IFN-k and -ω [279]. Virtually all nucleated cells can produce at least one type I interferon, affording a means of defense against viral infections and other pathogens; however, some cell types, namely plasmacytoid dendritic cells, produce larger quantities of type I interferons [280, 281]. Type I interferon production is triggered by various PAMPs, which are not limited to viral products; for example, activation of TLR4 by LPS can trigger type I interferon production by macrophages. The toll-like receptors, TLR3, TLR4, TLR7 (target of imiquimod), TLR8 and TLR9 are important inducers of type I interferon production [282].
The type I interferon receptor (IFNAR) is widely expressed on nucleated cells and is composed of IFNAR1 and IFNAR2 subunits and signals via JAK1, Tyk2, STAT1, STAT2 and interferon response factor (IRF) 9 [283]. Following IFNAR activation, a STAT1/STAT2 heterodimer forms and binds to the protein IRF9; this protein complex then translocates to the nucleus to regulate hundreds of interferon target genes. Upregulated interferon target genes include those encoding antiviral proteins with multiple mechanisms of action as well as MHC class I molecules, chemokines and other proteins. Activation of IFNAR has anti-proliferative and in some cases (e.g. keratinocytes), pro-differentiation effects [284]. Thus interferons limit propagation of virus by directly interfering with viral replication, limiting host cell proliferation and promoting presentation of viral antigens in the context of MHC class I to T cells [284]. Other effects of type I interferons include T cell, NK cell, macrophage and myeloid dendritic cell activation and promotion of T cells survival [23, 281].
The local and systemic actions of type I interferons have also been exploited therapeutically for dermatologic disease. For example, recombinant type I interferons are used to treat some cases of malignant melanoma and CTCL [285, 286]. Moreover, imiquimod, a TLR7 agonist, stimulates type I interferon production and is used topically to treat verruca vulgaris and molluscum contagiosum [287, 288]. Type I interferons are also implicated in the pathogenesis of psoriasis, lichen planus, cutaneous and systemic lupus, dermatomyositis, systemic sclerosis and sarcoidosis [23, 289–291]. In psoriasis, type I interferons derived from plasmacytoid dendritic cells and/or other cell types may initiate lesion formation through activation of myeloid dendritic cells that in turn produce IL-12 and IL-23 [23]. In support of this idea, psoriasis can develop or flare in patients treated systemically with type I interferons or topically with imiquimod [292, 293].
Type II Interferon (IFNγ)
Interferon gamma is a key mediator of many aspects of cutaneous and systemic immunity. In contrast to type I interferons, IFNγ production is limited to NK cells, T cell subsets (Th1 and CD8+ T cells) and ILC1 [294, 295]. Myeloid cell-derived cytokines including IL-12, IL-15 and/or IL-18 as well as IL-2 can stimulate production of IFNγ by T and NK cells [296, 297]. The IFNγ receptor, IFNGR, is a heterodimer of IFNGR1 and IFNGR1. In contrast to its ligand (IFNγ), IFNGR is widely expressed on a variety of cell types including keratinocytes, macrophages and dendritic cells [295]. Activation of IFNGR activates JAK1 and JAK2, and then, STAT1, which in turn regulates IFNγ target genes [295].
In contrast to type I interferons, IFNγ promotes MHC class II expression, is less critical for host defense against viruses and is more important for host defense against pathogens that target macrophages like Mycobacterial spp. [284, 295]. One of the critical functions of IFNγ in host defense is macrophage activation, a process in which activation of IFNGR leads to production of cytokines, nitric oxide and reactive oxygen species, which all contribute to clearance of intracellular pathogens that infect macrophages (i.e. Mycobacterial spp., as well as some fungi, parasites and other bacterial species) [298]. The critical role of IFNγ (and IL-12 which triggers IFNγ production) and macrophage activation in host defense against Mycobacterial spp. is highlighted by the fact that that human immunodeficiency disorders limited to type II interferon and/or IL-12 signaling (e.g. mutations in IFNGAR1, IFNGAR2, STAT1) greatly increase susceptibility to developing mycobacterial infections [298]. Other diseases in which IFNγ is implicated in pathogenesis include vitilgo, alopecia areata, allergic contact dermatitis, psoriasis and AD [23, 299–302].
A final note on IFNγ and its relevance to dermatology- interferon gamma release assays (IGRAs) are used to identify patients at risk for latent or active infections with Mycobacterium tuberculosis [303]. These tests involve incubating whole blood or PBMC with purified antigens from M tuberculosis; some T cells in the blood samples of patients with prior exposure to, or latent/active infection with M tuberculosis, can recognize these antigens and respond by producing IFNγ which is quantified [303]. Sufficiently high levels of IFNγ production in this assay alert the clinician to the possibility of latent or active infection. In addition to helping identify and treat patients at risk for infection, the results of this test help guide decisions regarding use of immunosuppressive medications for dermatologic and other disease indications.
Transforming Growth Factor β (TGF-β) Family
This family includes three isoforms of TGF-β (encoded by different genes) [304]. The isoform of TGF-β whose role in cutaneous immunology and disease is best understood is TGF-β1; hereafter though, all isoforms will be referred to collectively as TGF-β. In contrast to many cytokines described in this chapter, TGF-β and one or more of its receptors are expressed by most nucleated cells; sources of TGF-β in the skin include fibroblasts, macrophages and keratinocytes [305–308].
Like IL-1β and IL-18, TGF-β is synthesized in an inactive form and requires proteolytic cleavage to generate an active cytokine [309]. Unlike IL-1β and IL-18, maturation of TGF-β requires intracellular and extracellular processing (Fig. 15.4). Initially, the TGFB mRNA is translated into Pre-pro-TGF-β, which then undergoes proteolysis, removing the signal peptide and generating two proteins, a monomer of latency associated peptide (LAP) and a monomer of TGF-β; two LAP molecules form a homodimer, as do two TGF-β molecules, which then bind to each other (Fig. 15.4). Homodimers of LAP and TGF-β then bind another protein called latent TGF-β binding protein (LTBP) [309]. This complex is then secreted and can bind to the surface of cells bearing receptors for LAP or via binding of LTPB to extracellular matrix (ECM) components like collagen [310]. In this complex, TGF-β is inactive; depending on its location (i.e. ECM versus cell surface), proteolytic cleavage by a variety of different proteases or LAP receptor-mediated conformational changes liberate TGF-β, which can then bind and activate its receptors (Fig. 15.4) [310].
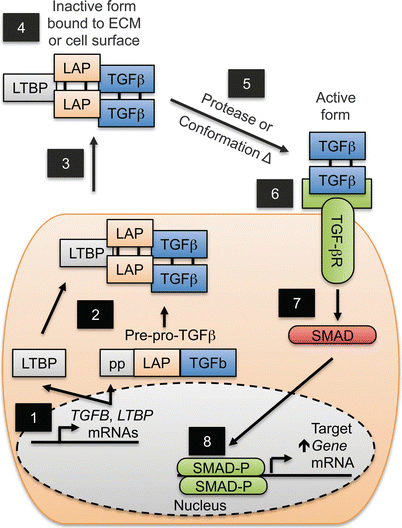
Fig. 15.4
Sequence of TGF-β production and receptor activation (TGF-βR): (1) TGFB and latent TGF-β binding protein LTBP mRNA is translated into Pre-pro-TGF-β and LTBP. (2) Pre-pro-TGF-β is cleaved into latency associated protein (LAP) and TGF-β followed by formation of a multi-protein complex of LAP, TGF-β and LTBP. (3) The complex is secreted. (4) It then binds extracellular matrix (ECM) components or LAP receptors on cells. (5) Proteolysis or conformational changes in the complex liberate TGF-β. (6) TGF-β then binds TGF-βR. (7) The TGF-βR then converts inactive SMAD proteins (SMAD) into an active phosphorylated form (SMAD-P). (8) Complexes of SMAD-P then translocate to the nucleus to regulate TGF-β target genes
There are three receptors for TGF-β designated I, II and III [304, 311]. The type III receptor binds to TGF-β but does not activate downstream signaling cascades [311]. Binding of TGF-β to the type II receptor recruits the type I receptor, triggering phosphorylation of the proteins SMAD2 and 3, which then recruit SMAD4 (Fig. 15.4). This complex in turn binds to and regulates the expression of TGF-β target genes [312].
While TGF-β has many functions, the focus here will be on wound healing and cutaneous immunity. In the skin and other organs, TGF-β regulates extracellular matrix (ECM) production by stimulating synthesis of collagen, fibronectin and integrins by fibroblasts; ECM degradation is also suppressed by TGF-β via reduced metalloproteinases production and increased synthesis of protease inhibitors [313]. These effects of TGF-β are thought to be important for ECM formation and preservation during the wound healing processes including scar formation. Excessive TGF-β activity is thought to contribute to hypertrophic and keloid scar formation and sclerotic or fibrotic disorders like systemic sclerosis (scleroderma) and nephrogenic systemic fibrosis [313–315]. Immunoregulatory effects of TGF-β include promotion of Treg, Th9 and Th17 cell development and inhibition of Th1 and Th2 cell differentiation (Fig. 15.1) [309]. In addition, TGF-β (along with IL-10 and IL-35) is an important effector molecule that regulatory T cells use to suppress effector T cells [316].
Chemokines
Chemokines are an entity unto themselves in structure, function and signaling mechanisms. They are critical regulators of leukocyte recruitment directing cell migration between secondary lymphoid tissues, the blood and peripheral tissues [317, 318]. The two largest subgroups of chemokines are designated CXC and CC, so named for the configuration of four conserved cysteine residues in these molecules. Chemokines activate G protein coupled receptors designated as CCR for CC chemokines and CXCR for CXC chemokines. Receptor engagement activates multiple signaling pathways that culminate in cytoskeletal reorganization, integrin activation and chemotaxis along a chemokine gradient [319]. Given similar mechanisms by which chemokines and their receptors act, this section will not discuss all chemokine and chemokine receptors used to recruit different leukocyte subsets to and from the skin, blood and lymph nodes; instead the role of chemokines in dendritic and T cell trafficking from and to the skin, respectively, will be presented as examples of how chemokines regulate leukocyte movement. In addition, Table 15.2 lists chemokines and chemokine receptors that promote chemotaxis of specific leukocyte subsets.
Table 15.2
Representative chemokines and receptors used by leukocyte subsets
Target cell | Chemokine receptor | Chemokine |
|---|---|---|
T cell | CCR4 | CCL17 (K, DC, E, M); CCL22 (K) |
CCR6 | CCL20 (K, E, M) | |
CCR8 | CCL1 (E, Ma) | |
CCR10 | CCL27 (K) | |
CXCR3 | CXCL9, 10, 11 (K, F, E, M) | |
CXCR4 | CXCL12 | |
B cell | CXCR4 | CXCL12 |
Monocyte | CCR1 | CCL3 |
CCR2 | CCL2 | |
CCR5 | CCL5, 8, 13 | |
CCR6 | CCL20 (K, E, M) | |
CXCR1 | CXCL6, 8 | |
CXCR2 | CXCL1 | |
CXCR3 | CXCL9, 10, 11 (K, F, E, M) | |
CXCR4 | CXCL12 | |
CX3CR1 | CX3CL1 | |
Eosinophil | CCR3 | CCL24, 26 |
Neutrophil | CCR1 | CCL3 |
CXCR1 | CXCL6, 8 | |
CXCR2 | CXCL1, 2, 3, 5, 6, 8 |
Dendritic Cell Trafficking from the Skin to Lymph Nodes
Sensitization to antigens encountered in the skin is dependent on acquisition of antigen by immature dendritic cells (see also Chap. 9) [317, 318]. Immature dendritic cells in the skin (and other sites) are adept at antigen uptake. Keratinocyte-derived CCL20 binding to CCR6 on Langerhans cells helps maintain this population of dendritic cells in the epidermis [318, 320, 321]. Antigen uptake and inflammation promote Langerhans cell maturation and migration to the skin from draining lymph nodes [318]. Inflammatory stimuli like TNF promote Langerhans cell migration out of the epidermis and into the dermis via downregulation of CCR6 and upregulation of CXCR4, the latter of which is engaged by the chemokine CXCL12 [318]. In the dermis, Langerhans cells and other dendritic cell subsets upregulate CCR7 that directs migration towards CCL19 and CCL21 produced in the lymph node [318, 322, 323]. In lymph nodes, mature dendritic cells present antigen to naïve T cells, promoting differentiation into effector memory T cells that mediate various aspects of host defense (Fig. 15.1) [317].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


