Causes
Frequency (%)
Idiopathic
40
Infection
22
Drug reaction
20
Connective tissue disease
12
Henoch-Schönlein purpura
10
Malignancy
<5
Systemic vasculitis
<5
Other systemic disease
<5
Initial Workup of Cutaneous Vasculitis
In cases of suspected vasculitis, the laboratory results and the histologic workup provide invaluable information beyond the history and the physical examination in determining the degree of systemic involvement and uncovering underlying causes for the vasculitic process. While physical findings can afford initial clues as to the types of vessels involved, the diagnosis of vasculitis can only be confirmed histologically. As the histopathologic diagnosis does not lend information regarding the degree of extracutaneous involvement, a clinic-pathologic correlation is necessary in the evaluation of these patients. Recommended initial laboratory workup for all patients and other tests that should be considered on a case-by-case basis have been described (Table 26.2) [13–15]. Taken together, these data ultimately contribute to decisions regarding prognosis and treatment modalities. For example, serum complement is a known mediator of vascular inflammation, and low levels indicate excessive consumption, suggesting more extensive or systemic involvement [16]. An elevated erythrocyte sedimentation rate (ESR) has been described in approximately 50 % of patients with limited cutaneous vasculitis [17]. An ESR of 40 mm/h or greater is associated with a high probability of systemic involvement [14].
Standard workupa | Other testsb |
|---|---|
CBC with differential | Renal biopsy |
CMP | Nerve conduction studies |
UA with microscopic evaluation | Hypercoagulability panel |
Biopsy for H&E and DIF | Echocardiogram |
Blood and urine cultures | |
Rheumatoid factor | |
C-reactive protein | |
ANA | |
ANCA | |
Cryoglobulins | |
Complement (C3, C4, CH50) | |
Hepatitis panel | |
Stool guaiac | |
Chest x-ray | |
HIV | |
SPEP/UPEP |
Skin biopsies sent for both routine hematoxylin and eosin (H&E) staining as well as direct immunofluorescence (DIF) are the cornerstones in determining whether physical findings represent true vasculitis or other conditions that can clinically mimic vasculitis. Decisions as to which lesions should be biopsied have direct impact on the diagnostic information elicited. Tissue should be acquired from lesions between 24 and 48 h after their appearance, as sampling before or after this time range may result in false-negative results. Biopsies with thrombosis or perivascular lymphocytic inflammation are characteristic of older lesions. In cases where suspicion of vasculitis is high but histology does not correlate with clinical findings, biopsy should be repeated. Likewise, if a medium-vessel vasculitis such as polyarteritis nodosa (PAN) is suspected, the biopsy must be of sufficient depth to include the subcutaneous tissue where these vessels are found. In general, ulcerated lesions should be avoided [1].
The diagnosis of vasculitis is confirmed unequivocally by the presence of an inflammatory infiltrate around and within the walls of vasculature with fibrin deposition. These areas of fibrinoid necrosis are accompanied by swelling and necrosis of endothelial cells, as well as secondary changes such as erythrocyte extravasation and necrosis leading to purpura and infarction, respectively [1, 12]. Apoptotic cells are seen frequently as well as overlying ulceration. Determination of vessel size, type of cellular infiltrate, depth, and degree of involvement on H&E stains helps in classification and generation of differential diagnoses.
The pathogenic features of vasculitis in the skin are related to vessel wall injury, which can be toxin mediated, immune mediated, or from direct infection, and all three mechanisms can result in the histologic pattern of fibrinoid necrosis. It is critical to identify those patients in which pathogenesis is caused by antibody-mediated toxicity and immune complex formation, as they are more likely to have extracutaneous involvement [1].
Deposition of immune complexes leads to complement activation, further recruitment of inflammatory cells and cytokines, and expression of adhesion molecules such as E-selectin, P-selectin, and intercellular adhesion molecule 1 (ICAM-1) [12]. With endothelial cell retraction, there is vascular deposition of immune complexes, neutrophil infiltration, edema, hemorrhage, and thrombosis [12].
Immunofluorescence is an essential diagnostic tool in the evaluation of cutaneous vasculitis, especially in the small-vessel group. This technique consists of the detection of immunoglobulins (Igs) and complement deposited within tissue. Deposition of IgA, IgG, IgM, and C3 in or around vessels identified by DIF characterizes antibody and immune complex–mediated vasculitis, and the patterns of deposition further classify disease. Lesion age is critical to evaluation, as up to 30 % of immune-mediated vasculitides are negative on DIF by 72 h and only C3 is detected after this point [17, 18]. Tissue samples are transported in Michel’s medium and stored at 4 °C prior to processing at specialized laboratories. Samples are washed, flash frozen, sectioned, and then stained to detect antibodies and complement in and around blood vessels. Certain diagnoses cannot be made without characteristic DIF patterns, which will be discussed with their associated conditions below.
Serologic testing has become routine in evaluation of vasculitis. In particular, antineutrophil cytoplasmic antibodies (ANCAs) have established clinical utility in dermatology [19]. Initially described in patients with rheumatoid arthritis, ANCA-associated vasculitides include small and medium-sized involvement such as Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, and many drug-induced cases of vasculitis, as well as systemic inflammatory conditions and connective tissue diseases [20]. It is believed that vasculitides associated with ANCAs have a distinct mode of pathogenesis [20, 21]. Antibodies can be directed against cytoplasmic (c-ANCA) and perinuclear (p-ANCA) neutrophil-derived products. C-ANCA antibodies are directed against proteinase 3 (PR3) and p-ANCA antibodies are directed toward myeloperoxidase (MPO), elastase and lactoferrin [20, 21]. Inflammatory cytokines are believed to induce the translocation of these targets to the surface of neutrophils, allowing binding of ANCAs and adherence to endothelial cells, ultimately causing damage to vessel walls [22]. Titers may predict clinical relapse or disease activity, and serial testing is recommended, as transient elevations in ANCAs can be seen with acute infections [23, 24]. Because c-ANCA is represented by only one antigen (PR3), enzyme-linked immunosorbent assay (ELISA) is a more sensitive and specific assay. Since several antigens are responsible for the p-ANCA pattern, immunofluorescence is preferred.
Drug–Induced Vasculitis
Medications from virtually every pharmacologic class (including herbal supplements) have been linked to drug-induced vasculitis, resulting in a range of clinical presentations (Table 26.3) [25–40]. In a MEDLINE database search for published cases of drug-induced vasculitis by ten Holder et al. [41], vasculitis was more often associated with propylthiouracil, hydralazine, colony stimulating factors, allopurinol, cefaclor, minocycline, D-penicillamine, phenytoin, isotretinoin, and methotrexate. The onset of findings after exposure to the causative agent is typically 5–20 days [1], and while withdrawal is often sufficient to reverse the vasculitic process, there have been cases of fatal drug induced allergic vasculitis in previously healthy patients [42]. There have also been reports of cutaneous vasculitis stemming from vaccines [43]. Ironically, many of the medications used for the treatment of systemic inflammatory conditions have also been linked to the development of cutaneous vasculitis [37–39]. Although the precise pathogenic mechanisms are varied, there appears to be a combination of cell-mediated and humoral immune responses contributing to the development of the observed vasculitis. As such, a thorough medication history is critical in the initial evaluation of the patient with suspected vasculitis, and all recently added medications should be discontinued. In those patients with both p-ANCA and c-ANCA, as well as eosinophilic infiltrates, the notion of drug-induced vasculitis should be entertained [44, 45]. Finally, one should keep in mind that the vast majority of drug-induced small-vessel vasculitis falls into the category of cutaneous leukocytoclastic angiitis (hypersensitivity vasculitis) or IgA vasculitis (Henoch-Schönlein purpura).
Table 26.3
Agents commonly reported
Medication | |
|---|---|
Minocycline | Famciclovir |
Carbamazepine | Montelukast |
Hydralazine | Infliximab |
Isoniazid | Etanercept |
Propylthiouracil | Rituximab |
Allopurinol | Ciprofloxacin |
Metformin | |
Vasculitis Associated with Systemic Conditions
Cutaneous vasculitis may result from a number of systemic triggers such as infectious, inflammatory, autoimmune, and malignant diseases, as well as pregnancy, and be of varying severity (Table 26.4) [46–51]. The workup of patients may reveal an underlying condition. Vasculitis can present prodromally or at any time during the disease. Among malignant diseases associated with vasculitis, hematologic cancers are seen most frequently. Bachmeyer and colleagues [52] found that of 95 hospitalized patients with hematologic malignancies, 23 (24 %) had biopsy proven cutaneous vasculitis. Skin findings developed before (26 %), during (39 %), and after (35 %) the diagnosis of malignancy. A thorough history, physical, and laboratory workup often point to the underlying disease processes, and in cases where there is failure to respond to treatment, investigation for occult malignancy should be considered.
IgA predominance?a | No IgA or IgG/IgM predominance?a |
|---|---|
IgA vasculitis | Non-IgA vasculitis |
Henoch-Schönlein purpura | Cryoglobulinemia II/III |
HUVSb | |
Rheumatoid vasculitis | |
Connective tissue disease | |
Wegener’s granulomatosis | |
Churg-Strauss syndrome | |
Microscopic polyangiitis | |
Behçet’s syndrome | |
Paraneoplastic vasculitis |
Treatment
If systemic conditions are excluded and potentially causative agents discontinued, treatment for vasculitis is driven by the severity of symptoms and extent of extracutaneous involvement. Many cases are isolated occurrences and can be managed supportively with rest, warming, compression and elevation of affected lower extremities. Symptomatic treatment for pain and inflammation can often be accomplished with nonsteroidal anti-inflammatory drugs, and pruritus with antihistamines. For recalcitrant disease or with evidence of systemic involvement, more aggressive therapy, including immunosuppressive agents, is necessary. Therapeutic decisions are based on the experience of the clinician and the details of the specific patient. Initially, systemic corticosteroids alone or in combination with steroid-sparing immunosuppressants are often employed. Long-term, steroid-sparing medications are used for control with systemic corticosteroids limited to disease flares, as to avoid the well known adverse effects of chronic corticosteroids. Each medication has certain adverse effects (i.e., bone marrow toxicity, impaired renal function and drug-induced bladder complications) and regular and frequent laboratory evaluations are necessary to screen for these. Corticosteroids, cyclophosphamide, azathioprine, methotrexate, colchicine, dapsone and mycophenolate mofetil have been reported to treat vasculitis [14].
Corticosteroids demonstrate great utility across the spectrum of autoimmune and inflammatory disorders by suppressing effects of cytokines (IL-1, TNF-α), T-cells and B-cells. Circulating T-cell depletion occurs from corticosteroid enhanced circulatory emigration, induction of apoptosis, inhibition of T-cell growth factors, and impaired release of cells from lymphoid tissues [53]. Higher corticosteroid doses render significant B-cell effect and reduced immunoglobulin production.
Cyclophosphamide is an alkylating agent first developed to treat malignancies and has been used in the treatment of certain autoimmune diseases. Having potent immunosuppressive effects it depresses B-cell function more than T-cell function. Cyclophosphamide is used often in combination with corticosteroid therapy, considered the first-line treatment for Wegener’s granulomatosis [54]. Cyclophosphamide is also used for remission induction with less toxic medications (i.e., methotrexate or azathioprine) for maintenance therapy.
Azathioprine, initially used in transplantation medicine for immunosuppressant properties, is also often used for its anti-inflammatory qualities. Azathioprine’s active metabolites are 6-thioguanine monophosphate and other metabolites [55]. The exact mechanisms by which these purine analogs lead to immunosuppressive and anti-inflammatory effects are not clear.
Methotrexate is an antimetabolite chemotherapeutic agent with immunosuppressive effects used for inflammatory and immune-mediated processes. This drug is a potent competitive antagonist of dihydrofolate reductase, thereby inhibiting DNA synthesis and cell division. The immunosuppressive effects of methotrexate stem from inhibition of DNA synthesis in immunologically competent cells, suppressing antibody responses [56]. The anti-inflammatory effects are predominantly mediated by adenosine [57].
Colchicine has both antimitotic and anti-inflammatory properties and has demonstrated utility in dermatologic diseases characterized by polymorphonuclear leukocyte infiltration. Within leukocytes, colchicine binds to the dimers of tubulin, preventing the assembly of tubulin subunits into microtubules, thereby leading to arrest of mitosis [58].
Dapsone is useful in treating many skin diseases, primarily those characterized by neutrophilic infiltrates in the skin. Dapsone has been thought to accomplish this by altering neutrophil chemotaxis and respiratory burst, thereby leading to reduced oxidative damage in tissues [59]. The exact mechanisms are not clear, however.
Mycophenolate mofetil has demonstrated efficacy in multiple types of autoimmune and inflammatory skin diseases. MMF is a prodrug which first must be converted to the active drug form mycophenolic acid (MPA). MPA inhibits inosine monophosphate dehydrogenase, thus depriving the T- and B-lymphocytes of purine metabolites necessary for growth and replication and subsequent immunosuppression [60].
Often, these treatment regimens are sufficient to control disease activity with few adverse effects when appropriate monitoring is performed. Some patients, however, may be refractory to these treatments or develop intolerable side effects from therapy. These factors have ushered in an era of alternative treatment modalities with less toxicity and greater efficacy. More recently, effective treatments with tumor necrosis factor-α (TNF-α) inhibitors [61, 62] and anti-CD20 antibodies have been described [63, 64]. Rituximab is a chimeric monoclonal antibody directed against the B-cell lineage specific CD20 antigen, yielding B-cell depletion activity and subsequent inhibition of antibody production. Originally developed for the treatment of B-cell non-Hodgkin’s lymphoma, rituximab has increasingly been used to treat a variety of immune-mediated disorders.
Clinical Mimickers of Cutaneous Vasculitis
A variety of conditions are capable of clinically simulating cutaneous vasculitis and have been termed pseudovasculitides [65]. Many of these conditions can be excluded by biopsy, and are typically associated with conditions that cause hemorrhage or vessel occlusion, and should always be in the differential diagnosis for vasculitis. Vessel wall dysfunction or incompetence from infiltrative processes, nutritional deficits such as scurvy [66], infection, embolism, vasospasm, and trauma can all present with varying degrees of purpura, petechiae, ecchymoses, ulcers, and necrosis. Likewise, hypercoagulable states such as antiphospholipid syndrome and factor V Leiden can lead to similar clinical pictures and need to be excluded [8].
Classification of Cutaneous Vasculitis
Classification of vasculitis in the skin is typically based on the size of predominantly affected blood vessels and type of inflammatory response, which when interpreted with DIF examination and laboratory workup, correlate with disease etiology and affect the treatment decisions. Vessels of varying sizes are frequently involved, however, as vasculitic processes do not always recognize arbitrary boundaries of vessel size. Most texts refer to the classification schemes outlined by the Chapel Hill Consensus Criteria (Table 26.5) or the American College of Rheumatology, although it is often difficult to characterize individual patient variations [67–69].
Table 26.5
Classification of cutaneous vasculitis according to the Chapel Hill Consensus Conference [67]
Large vessel vasculitis | Medium vessel vasculitis | Small vessel vasculitis |
|---|---|---|
Takayasu arteritis | Polyarteritis nodosa | Microscopic polyangiitis |
Giant cell arteritis | Kawasaki disease | Granulomatosis with polyangiitisa |
Eosinophilic granulomatosis with polyangiitisb | ||
Cryoglobulinemic vasculitis | ||
IgA vasculitisc | ||
HUVS (anti-C1q vasculitis)d |
Small blood vessels are ubiquitous in the skin. They include arterioles, capillaries, and postcapillary venules. They are typically 50 μm or less in diameter, and may not have a fully developed muscular layer. Clinical lesions of cutaneous small-vessel vasculitis (CSVV) are most commonly nonblanchable and purpuric, and are found in dependent areas (buttocks, back, lower extremities). When urticarial lesions are present, they are less pruritic, short-lived (<24 h), and can occur anywhere on the body. They include Wegener’s granulomatosis, Churg-Strauss syndrome, microscopic polyangiitis, Henoch-Schönlein purpura, essential cryoglobulinemic vasculitis, and cutaneous leukocytoclastic angiitis.
Medium-sized blood vessels are larger than 50 μm, have fully developed muscular layers, and are located deeper within the dermis or subcutaneous fat. Clinically, these processes present with livedo, nodules, ulcerations, or digital infarcts. Wedge biopsy is typically needed for sufficient diagnostic yield, and biopsy of necrotic or ulcerated areas is of low yield. Included among this group are polyarteritis nodosa and Kawasaki disease. Vasculitis of these vessels is commonly referred to as necrotizing vasculitis, reflecting the hyalinization, coagulative necrosis, and degeneration of muscular layers, where it is more readily visible. Occasionally nerve or muscle biopsy can provide additional diagnostic information if histology is inconclusive.
Large-vessel vasculitides rarely have cutaneous manifestations and include giant cell (temporal) arteritis and Takayasu’s arteritis, and will be mentioned only briefly.
Cutaneous Leukocytoclastic Angiitis
As defined by the Chapel Hill Criteria, cutaneous leukocytoclastic angiitis (CLA) is the term applied to patients with hypersensitivity vasculitis. Exogenous chemicals, infectious agents, cytokines, and circulating immune entities that do not strongly activate complement can cause CLA by inducing an inflammatory cascade in endothelium of CSVV. Most commonly triggered by infections or drugs, the onset is acute with both palpable and nonpalpable purpuric and urticarial lesions on the lower extremities appearing 5–20 days after initial exposure, and 2–4 days after repeat exposures (Fig. 26.1) [70]. These cases tend to be single episodes, and relapsing cycles can result from systemic inflammatory conditions, infection, and malignancy. Extracutaneous involvement is rare, with the exception of constitutional symptoms caused by mediators of inflammation released locally [71]. Serum sickness resulting from the injection of nonhuman serum can present with a similar picture, but is rarely seen in modern practice.
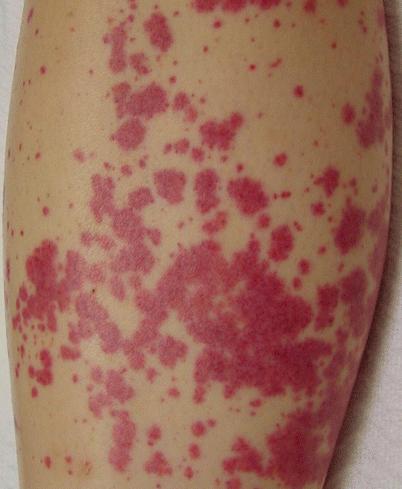
Fig. 26.1
Acute palpable purpura of cutaneous leukocytoclastic angiitis (hypersensitivity vasculitis)
Routine laboratory tests are usually normal, as extracutaneous disease is rare. The ESR is elevated in up to 50 % of cases, while complement levels and urinalysis are normal. There are no specific serologic markers for CLA, making it largely a diagnosis of exclusion, and normocomplementemic urticarial vasculitis is likely to be a clinical variant of this condition [72].
On histology, there is a neutrophil predominant vasculitis of superficial vessels with varying numbers of surrounding eosinophils (Fig. 26.2). Direct immunofluorescence is positive in roughly half of these biopsies, displaying mild to moderately intense granular IgM deposits with weak or absent C3. The lack of complement involvement may correlate with the relatively benign course of this condition and low level of systemic involvement. Although DIF is frequently negative, it is a key factor in discriminating CLA from IgA vasculitis, which shares the same triggers and clinical presentation [73, 74].
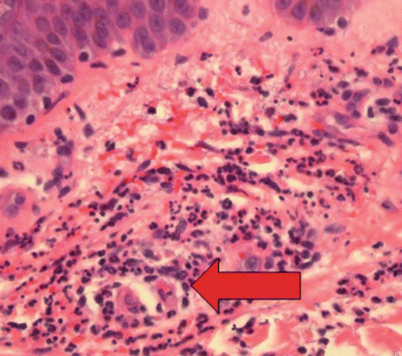
Fig. 26.2
Hematoxylin and eosin (H&E) staining of skin biopsy from a patient with cutaneous small-vessel vasculitis. This is a leukocytoclastic vasculitis involving small, superficial vessels, rich in neutrophils (arrow)
Up to half of the cases of CLA are idiopathic and resolve spontaneously. In cases with a known trigger, treatment consists of removal of the offending agent or resolution of underlying systemic condition. Immunosuppressive treatment is largely unnecessary in CLA, with the exception of the most severe cases, and is aimed at reducing constitutional symptoms and synovitis. Moderately dosed corticosteroids (0.5 mg/kg/day) are a reasonable option until symptoms resolve. Recalcitrant cases warrant more extensive investigation. Rituximab therapy has demonstrated efficacy in the treatment of cutaneous angiitis refractory to high dose corticosteroids and cyclophosphamide [75].
Henoch-Schönlein Purpura
Henoch-Schönlein purpura (HSP) is defined as an IgA-mediated syndrome presenting with a tetrad of purpura, abdominal pain, arthralgias, and hematuria [76]. It is most commonly seen in children, but there have been increasing reports of HSP in adults [76, 77]. HSP runs a more severe course and is more likely to cause long-term renal disease in adults compared to children [78, 79].
As with CLA, HSP is often preceded by medications or infection, most commonly upper respiratory, gastrointestinal (GI), and genitourinary (GU). When associated with GI and GU infections, it can be difficult to discern findings related to infection from those caused by the vasculitic process itself. Approximately 50 % of those affected develop systemic involvement such as nephritis, neuropathy, and GI symptoms. [80] Skin lesions are seen in all patients and are typically palpable purpura or urticaria of the lower extremities and buttocks that turn into purpura with annular configuration (Fig. 26.3). Koebnerization is known to occur in HSP. A subset of patients displays only urticarial lesions, and lesions above the waist have been associated with renal disease, as are elevated ESR, fever, and adult onset [81]. Women have an increased risk for the development of proteinuria or preeclampsia in future pregnancies [82, 83].
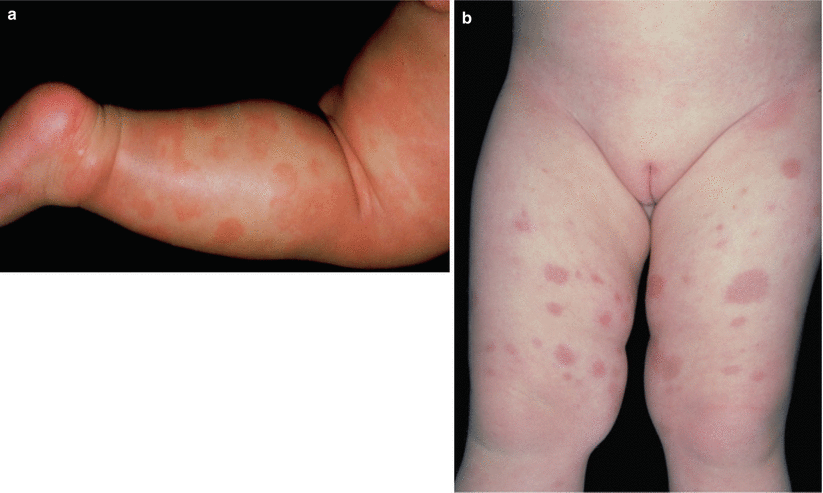
Fig. 26.3
(a, b) Infants with typical lesions of Henoch-Schönlein purpura. Lesions above the waistline are associated with a worse prognosis (Courtesy of Dr. M. Mercurio)
Extracutaneous involvement can appear in any organ, with the kidneys, GI tract, and joints being most common. Renal failure secondary to glomerulonephritis is the most serious complication of HSP.
Renal involvement occurs in about 33 % of children and 63 % of adults with HSP. [84] The risk of progression to renal insufficiency ranges from 5 to 15 % in children and seems to be much higher in approximately 30 % in adults [77]. Children with renal involvement have a higher incidence of hypertension and renal failure as adults, and 15 % of children on hemodialysis have renal failure secondary to HSP. [85] Urinalysis is an absolute requirement for the patient with suspected HSP, with hematuria providing the most sensitive measurement for renal involvement. Recent studies have revealed abnormalities of IgA1 glycosylation and formation of autoantibodies to aberrantly glycosylated IgA1 molecules with subsequent mesangial deposition and renal disease [86, 87]. New noninvasive disease activity markers (aberrantly glycosylated IgA1 and its immune complex) may aid in the diagnosis and help guide therapy. Gastrointestinal involvement can range from pain to hemorrhage and necrosis of the bowel from mesenteric vasculitis, and pulmonary hemorrhage has also been reported [88]. However, it is difficult to determine if involvement of these organs is the result of a prodromal infection or from the vasculitic process itself.
There is mounting evidence that acute hemorrhagic edema of infancy (AHEI) is a variant of HSP, although in contrast with HSP, extracutaneous involvement is rare and IgA deposition is rarely seen with DIF. [89] AHEI is frequently seen after bacterial infection, and lesions appear on the face and extremities. There are no specific serology associations with HSP, although up to 40 % of adult patients demonstrate some degree of IgA gammopathy [81]. When appropriate, additional workup should include serum and urine protein electrophoresis (SPEP and UPEP), total immunoglobulin quantitation, and immunofixation.
Routine H&E staining reveals a neutrophil-rich small-vessel vasculitis of the superficial dermis with leukocytoclasia and few eosinophils (Fig. 26.2). DIF provides the gold standard for diagnosis, with IgA deposits in small, superficial vessels found in virtually all biopsies. Granular deposits of other Ig classes are occasionally seen, with IgA being the most prominent [87].
As with any of the vasculitides, treatment decisions hinge on the degree of involvement and constitutional symptoms. While the cutaneous findings are largely refractory to therapy, synovitis and GI symptoms are quite responsive to moderate doses of oral corticosteroids. Skin lesions frequently have an initial response to steroids, followed by rapid relapse upon completion of treatment. Antimetabolites such as azathioprine and mycophenolate mofetil, as well as alkylating agents can be effective for treating glomerulonephritis. However, there is little evidence that any other immunosuppressive therapy beyond corticosteroids is effective in HSP nephritis [90].
Urticarial Vasculitis
Urticarial vasculitis presents clinically as urticaria of the trunk and proximal extremities, and histologically as vasculitis (Fig. 26.4). It occurs in two forms differentiated by serum complement levels. The normocomplementemic variant is a subset of CLA, while the hypocomplementemic type (HUVS) is a subset of systemic lupus erythematosus (SLE) [72, 91]. Patients with low levels of C3, C4, and total serum hemolytic complement (CH50) have dramatically increased rates of complement consumption. CH50 is a more sensitive predictor of HUVS since C3 and C4 are acute-phase reactants and may be normal in mild disease or early-stage disease. Thorough workup of patients with urticarial vasculitis should include the quantitation of C3a, C5a, and C3bi when available [72, 74].
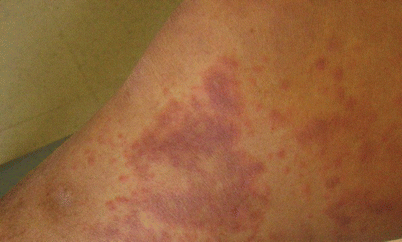
Fig. 26.4
Urticarial vasculitis. These lesions are difficult to distinguish clinically from traditional urticaria and can occur on the face and upper extremities (Courtesy of Dr. F. Tausk.)
Ninety percent of normocomplementemic urticarial vasculitis (NUV) are triggered by infection or medications, and are self-limited without indication of systemic involvement [73]. The clinical differential includes urticarial HSP, neutrophilic urticaria, Schnitzler’s syndrome, and urticarial cryoglobulinemia II and III.
Neutrophilic urticaria, also known as polymorphonuclear predominant urticaria (PPU) is a subset of chronic urticaria [92]. Lesions are typically pruritic, and mild constitutional symptoms can be present. Histologic features of PPU can be confused with NUV, as perivascular neutrophilic infiltrates can be dense, with occasional karyorrhexis; however, there is no definite disruption of vasculature and DIF is consistently negative in PPU. Chronicity and no obvious underlying cause argue against NUV. Treatment consists of antihistamines, leukotriene antagonists, dapsone, or colchicine. In cases of PPU it is important to rule out Schnitzler’s syndrome, which is characterized by NUV, fever, lymphadenopathy, hepatosplenomegaly, peripheral neuropathy, bone pain, and monoclonal IgM gammopathy [93]. In contrast to patients with NUV, the hypocomplementemic variant of urticarial vasculitis presents as a chronic, relapsing syndrome (HUVS) with signs and symptoms typically seen with SLE, including fever, arthralgias, and myalgias [73, 91].
In contrast to lesions of NUV, HUVS lesions tend to have a purpuric component upon careful examination. Synovitis, GI involvement, and scleritis are not uncommon associated findings. A subset of HUVS patients with more overt symptoms of SLE will display angioedema, thought to be mediated by a high rate of consumption of C1 esterase caused by autoantibodies to C1q. In both variants of urticarial vasculitis, histology reveals a dense neutrophilic infiltrate in and around the walls of small vessels that disrupts normal architecture. In HUVS, dermal edema and neutrophils extend to the dermal-epidermal junction (DEJ), resulting in vacuolar changes and clefting of the basement membrane zone (BMZ). The only serologic marker associated with NUV is an elevation in ESR in up to 70 % of patients, while essentially all patients with HUVS have high ESR and an antinuclear antibody (ANA) titer greater than 1:320 at some point in their disease course [1].
As with CLA, 50 % of patients with NUV have immune deposition within and around blood vessel walls seen with DIF. IgM is seen more frequently than IgG, and C3, if present, is weak and patchy. In contrast, essentially all patients with HUVS show significant IgG and C3 intravascularly and perivasculary within superficial dermal vessels extending to the BMZ (Fig. 26.5). It has been reported that this latter finding resembles the lupus band test, further linking HUVS with SLE [74, 94].
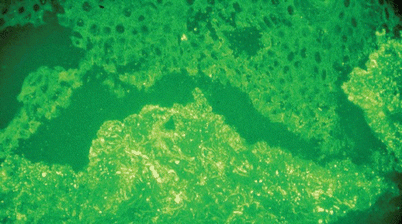
Fig. 26.5
Direct immunofluorescence of biopsy from a patient with hypocomplementemic urticarial vasculitis syndrome (HUVS) revealing granular immunoglobulin G (IgG) in and around superficial vessels
Treatment of NUV is similar to that for CLA— conservative and directed toward alleviation of mild symptoms. HUVS, however, frequently requires systemic immunosuppressive therapy such as corticosteroids, azathioprine (3–4 mg/kg/day), or mycophenolate mofetil (40–50 mg/kg/day); these medications are usually sufficient. Anti-inflammatory agents such as dapsone, colchicine, methotrexate, and calcineurin inhibitors are not typically effective, as they have no effect on the production of immune complexes or anti-C1q synthesis.
Cryoglobulinemia
Cryoglobulinemic vasculitis affects both small and medium-sized skin blood vessels. Cryoglobulins are antibodies that precipitate with cold, and three types of cryoglobulinemia exist as defined by the type of antibodies present. Type I is a monoclonal gammopathy that commonly presents as a hyperviscosity syndrome and thrombotic events in the context of myeloproliferative disorders, and does not represent true vasculitis. Type II is mixed, with an IgM monoclonal component (usually an IgM rheumatoid factor) in conjunction with polyclonal gammopathy. Type III consists of polyclonal cryoglobulins. Because types II and III readily form immune complexes, they are more likely to cause vasculitis, as opposed to the thrombotic vasculopathy seen with type I cryoglobulins [1, 95, 96]. Cryoglobulinemia type II has antibodies that form immune complexes with much higher avidity and levels of complement fixation, resulting in more significant clinical syndromes. Hepatitis C is by far the most common cause of type II cryoglobulinemia, representing essentially all cases previously labeled as essential mixed cryoglobulinemia (Fig. 26.6) [95, 96]. It is felt that the virus stimulates the immune system chronically, causing B-cell expansion and production of autoantibodies. Other associated infections include endocarditis, hepatitis B, and HIV. Connective tissue disease, other autoimmune conditions, and malignancy have been associated with both types II and III cryoglobulinemic vasculitis [97].
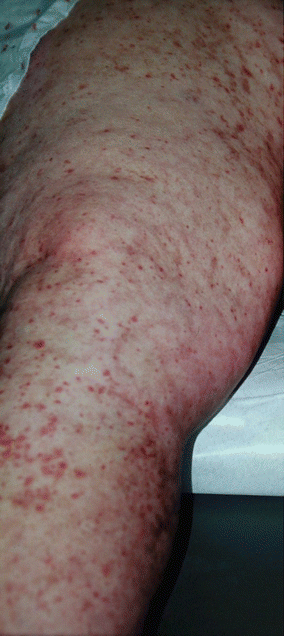
Fig. 26.6
Clinical image of patient with known hepatitis C presenting with the CSVV (purpura) and medium-sized vessel vasculitis (livedo) lesions associated with cryoglobulinemia type II
Clinically, skin lesions associated with cryoglobulinemia type I are indistinguishable from those seen with type II, and resemble the palpable purpura and urticarial findings seen with any CSVV. As with most vasculitides other than urticarial vasculitis, lesions most commonly appear on the lower extremities. Cryoglobulin-associated nonpalpable purpura can also present as the capillaritic eruption seen in Schamberg’s disease. The benign hypergammaglobulinemic purpura seen in Waldenström’s (lymphocytic vasculitis) is clinically, serologically, and histologically indistinguishable from that seen with cryoglobulinemia type III, and, as such, is generally believed to belong to the same spectrum of disease [97]. Pigmented purpura above the waistline or involving the soles of the feet favor cryoglobulinemic vasculitis over Schamberg’s, as do lesions seen at different stages of evolution, ulcerative lesions, and constitutional symptoms. Ulceration signals the involvement of medium-sized vessels and can help differentiate from other CSVV. As mentioned, medium-sized vessels can also be seen with cryoglobulinemia.
These patients also tend to have systemic symptoms such as nephropathy, neuropathy, arthralgias, and gastrointestinal involvement [1, 98].
Serologically, cryoglobulin type II patients demonstrate monoclonal IgM rheumatoid factor (RF) and polyclonal IgG. All patients have an RF greater than 1:320, and over 90 % have decreased C4 but essentially normal C3. Therefore, a negative cryoglobulin assay and negative RF in the setting of low C4 virtually excludes cryoglobulinemia type II, whereas a low titer cryoglobulinemia in the absence of vasculitis is common after many infections. A positive cryoglobulinemia with negative RF activity likely represents an incidental finding and not vasculitis. Cryoglobulinemia type III demonstrates a polyclonal gammopathy without any specific monoclonal spike, and low levels of both C3 and C4 [1].
Histology demonstrates findings consistent with CSVV or both CSVV and medium-sized vessel vasculitis (MSVV), but not MSVV alone. Some texts report the deposition of a nonspecific, homogeneous intravascular infiltrate associated with cryoglobulinemic vasculitis; however, this likely represents thrombotic vasculopathy seen with type I cryoglobulinemia and not true vasculitis [97].
Direct immunofluorescence in cryoglobulinemia type II usually reveals significant granular IgM and C3 deposition in and around small and medium sized vessels, while type III has both IgG and IgM in addition to C3. In practice, it is often difficult to distinguish between types II and III cryoglobulinemia by DIF.
Because types II and III cryoglobulinemias are difficult to distinguish histologically and clinically, treatment decisions should hinge on the serologic workup and be directed toward the underlying etiology (e.g., hepatitis C) as well as the vasculitis. Treatment of the latter is similar to that of other vasculitides and is based on combinations of steroids and steroid-sparing agents that are used in an effort to target antibody and immune complex–mediated inflammation. Differences do exist, however, when treating the underlying condition in type II as opposed to type III cryoglobulinemia. For instance, in the setting of hepatitis C–induced type II cryoglobulinemia with high titers of cryoglobulins and RF and low complement, antiviral treatment with interferon-γ could potentially result in massive formation of immune complexes, exacerbation of vasculitis, and multi-organ failure [98]. This group of patients should be treated with immunosuppressive agents of low hepatotoxicity for at least 6 months prior to antiviral therapy [99].
An additional difference in the management of type II versus type III cryoglobulinemia is with plasmapheresis aimed at removing pathogenic immunoglobulins. Because antibodies in type II cryoglobulinemia are intravascular as opposed to the intra- and extravascular deposition seen in type III cryoglobulinemia, plasmapheresis is much more effective in the prior condition, as extravascular antibodies are not affected by plasmapheresis. Rituximab is a newer therapeutic option that has demonstrated its utility by its B-cell depletion activity and subsequent inhibition of antibody production such as that which occurs in cryoglobulinemia. Studies have demonstrated the efficacy of rituximab as monotherapy in the treatment of cryoglobulinemia vasculitis, or when used in combination with corticosteroids [100]. Rituximab plus corticosteroids appears to provide better efficacy in patients with relapsing disease or disease refractory to other therapies [101, 102].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


