, Camille E. Introcaso3, Stephen K. Richardson4 and Alain H. Rook5
(1)
Department of Dermatology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA
(2)
Department of Dermatology, Hospital of the University of Pennsylvania, Philadelphia, PA, USA
(3)
Department of Dermatology, Pennsylvania Centre Dermatology, Philadelphia, PA, USA
(4)
Department of Dermatology, Tallahassee Memorial Healthcare Hospital, Dermatology Associates of Tallahassee, Tallahassee, FL, USA
(5)
Department of Dermatology, Perelman School of Medicine at the University of Pennsylvania, 3400 Civic Center Boulevard, Building 421, Philadelphia, PA 19104, USA
Abstract
Cutaneous T-cell lymphomas (CTCLs) are a group of extranodal non-Hodgkin’s lymphomas (NHLs) that present primarily in the skin. The most common type of CTCLs, mycosis fungoides (MF) and Sézary Syndrome (SS), were first described over two centuries ago, and since that time, the clinical characteristics, pathophysiology, and immunobiology have been characterized in detail. Derived from skin-homing, mature, effector T-cells that usually express CLA/CD4/CCR4/CCR10 and lack T-cell markers CD7 and/or CD26, the malignant MF/SS cells typically have a Th2 phenotype. With more advanced disease, Th2 cytokines predominate and result in decreased host cell-mediated immunity that likely contributes to increased susceptibility to infection and disease progression. Early aggressive systemic chemotherapy has not been shown to improve overall survival in MF/SS and over the past 40 years [1] the therapeutic approach has undergone a paradigm shift, such that skin-directed therapies (SDTs) and systemic immune-modifying biologics play a central role in initial MF/SS management. This chapter will review MF/SS clinical presentation, staging work-up, pathophysiology, immunobiology, and how these have shaped current treatment strategies. In particular, MF/SS chemokine biology, immune defects, and immune modifying therapies, including the new frontier of hematopoietic stem cell transplantation will be specifically highlighted.
Keywords
Cutaneous T-Cell LymphomaCTCLNon-Hodgkin’s lymphomasNHLsMycosis fungoidesMFSézary SyndromeSSDermatitisPsoriasisParapsoriasisSkin conditionSkin diseaseTumor-node-metastasis-bloodTNMBClinical Presentation
CTCLs are a heterogeneous group of extranodal lymphomas and were the subject of a new joint classification system in 2005 by the World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) (Table 41.1) [2]. MF/SS is relatively uncommon and comprises approximately 3.9 % of all NHLs [3]. The incidence of CTCL has increased since 1973 with an annual age-adjusted incidence of 6.4–9.6 cases per million people in the United States [4]. The cause of the vast majority of MF/SS cases are unknown, despite numerous environmental and infectious etiologies having been investigated as potential sources of chronic antigenic stimulation. These include human T-cell lymphotrophic virus (HTLV), cytomegalovirus (CMV), Borrelia burgdorferi, human herpes virus-8 (HHV-8), Staphylococcus aureus, Chlamydia species, and chemical exposures such as aromatic or halogenated hydrocarbons [5, 6].
Table 41.1
EORTC-WHO classification of cutaneous T- and NK/T-cell lymphomas (2005)
Mycosis fungoides and variants/subtypes |
|---|
Sezary syndrome |
Primary cutaneous CD30+ lymphoproliferative disorders |
Subcutaneous panniculitis-like T-cell lymphoma |
Extranodal NK/T-cell lymphoma, nasal type |
Adult T-cell leukemia/lymphoma |
Primary cutaneous peripheral T-cell lymphoma, unspecified |
Aggressive epidermotropic CD8+ T-cell lymphoma |
Cutaneous γδ T-cell lymphoma |
PC CD4+ small/medium-sized pleomorphic T-cell lymphoma |
Peripheral T-cell lymphoma, other |
MF classically presents as scaly oval or annular patches/plaques (T1-2 tumor designation) on sun-protected areassuch as the trunk, buttocks, axillae, groin, and proximal extremities, (“bathing trunk” distribution) with or without pruritus (Fig. 41.1a–c). Typically, it has indolent behavior and in early disease when host immunity is intact and lesions may wax and wane spontaneously. Because of this, early disease may take years before definitive diagnosis is made and skin biopsies may demonstrate non-specific dermatitis. Lesions may also less commonly affect sun exposed areas such as the face and can also result in localized hair loss (as seen in the follicular variant). They may also progress to thicker, more infiltrated plaques (Fig. 41.1c), ulcerated tumors (T3 disease) (Fig. 41.1d), or coalesce into a confluent erythema >80 % body surface area (“erythroderma” or T4 disease) (Fig. 41.1d). In these cases, the disease can cause more symptoms and behave more aggressively.
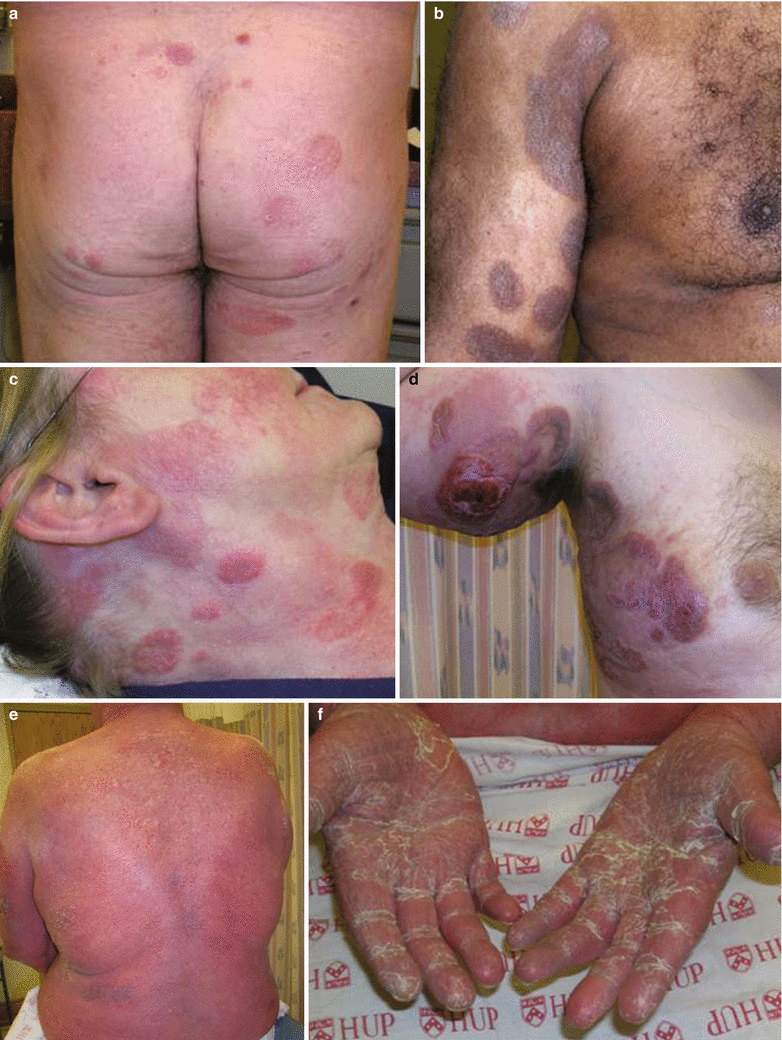
Fig. 41.1
Skin lesions in mycosis fungoides (MF)/Sézary syndrome (SS). Oval patches of MF in typical sun-protected areas of the buttocks (a) and proximal arms/axillae (b). (c) Raised, annular plaques of MF. (d) MF plaques and an ulcerated tumor. SS patient with erythroderma (e) and palmar keratoderma (f)
In addition to the classic presentation, MF has myriad other clinical morphologies and histological variants (classic Alibert-Bazin subtype, erythrodermic/Sezary Syndrome, unilesional, hypopigmented, pagetoid reticulosis, follicular/follicular mucinosis, syringotropic, granulomatous/slack skin, bullous/vesicular, palmoplantar, pigmented purpuric dermatosis-like, interstitial, icthyosiform, hyperkeratotic/verrucous, vegetating/papillomatous) [7] and has been referred to as one of the great clinical imitators, similar to cutaneous syphilis and sarcoidosis. Subtypes may be characterized by more indolent behavior (hypopigmented MF) or by more aggressive behavior (follicular, granulomatous), depending on their clinical course. Follicular MF, even with limited lesions, can be recalcitrant to therapy given the depth of the malignant infiltrate.
SS typically has a more aggressive course than MF from its onset, and typically presents as de novo erythroderma with leukemic peripheral blood involvement by the atypical, hyperconvoluted, cerebriform, malignant T-cells known as Sézary cells (also previously referred to as Lutzner cells or cellules monstrueuses). This is in contrast to patients who have MF that evolves into erythroderma over time (erythrodermic MF). Originally, SS was described with the clinical triad of erythroderma, lymphadenopathy, and palmoplantar keratoderma (Fig. 41.1e). SS patients may also have severe pruritus, active scaling/desquamation, prominent conjunctival eversion of the eyelids (referred to as ectropion), diffuse alopecia, chills, low grade fevers, night sweats, or fatigue.
MF/SS patients, particularly erythrodermic patients, are heavily colonized by skin bacteria and are susceptible to recurrent infections with correct Staphylococcus aureus, which often are methicillin-resistant (MRSA). Patients with ulcerated tumors are also at high risk for bacterial sepsis and in general, infections have previously been the leading cause of mortality in MF/SS patients [8].
MF/SS patients may also present with other concomitant skin conditions, such as the chronic, recurrent primary cutaneous CD30+ lymphoproliferative disorders (LPDs) such as lymphomatoid papulosis (LyP) or CD30+ primary cutaneous anaplastic large cell lymphoma (ALCL) [9]. Patients with MF/SS may also be at higher risk for having secondary malignancies, such as other skin cancers (nonmelanoma and melanoma) and secondary lymphomas [10]. Young patients with MF/SS diagnosed before 30 years of age may experience an increased risk of secondary malignancies, particularly melanoma and lymphoma, but, despite this risk, have a favorable prognosis [11].
Diagnosis
Early diagnosis of MF can be elusive, as the clinical and histopathology of early MF lesions can mimic benign dermatoses, such as eczematous dermatitis, allergic contact dermatitis, psoriasis and parapsoriasis. Traditionally, the diagnosis is made when a patient presents with typical lesions and skin biopsy reveals atypical lymphocytes (with large, hyperchromatic and hyperconvoluted nuclei) within the epidermis, in the absence of epidermal spongiosis, and often associated with epidermal dendritic cells (DCs) to form the pathognomonic “Pautrier’s microabcesses.” Currently there are sophisticated laboratory tools to aid in diagnosis, which include tissue immunohistochemistry and flow cytometry (which can demonstrate CD3 expression, CD4 > CD8 expression of greater than 4:1, as well as characteristic T-cell lineage antigen loss of CD5, CD7, CD26, and less often CD3). Molecular detection of clonal T-cell receptor (TCR) gene rearrangements (β and γ chains) in the skin or other tissue (such as the blood or lymph node) can also further strengthen the diagnosis [12]. These days, the standard evaluation of early MF often involves assessment of multiple criteria (clinical, histopathological and molecular) [13]. However, it must be noted that T-cell clonality can also be detected in benign and autoimmune inflammatory dermatoses and should not be automatically equated with neoplasia [14, 15]. Diagnosis ultimately rests on accurate clinicopathologic correlation with adjunctive molecular studies, if appropriate.
In SS, leukemic blood involvement was traditionally measured by the Sézary prep, visual examination of a peripheral blood buffy coat smear for the atypical Sézary cells (Sézary count >5 % total lymphocytes was considered significant). However, this method was laborious and was subject to considerable inter-observer variability. More sensitive and specific tools to measure peripheral blood involvement include anti-TCR Vβ-specific monoclonal antibodies, or T-cell surface marker antibodies (CD3, CD4, CD7, CD8, CD26) using fluorescence-activated cell-sorting (FACS) analysis/flow cytometry.
The identification of a clonal malignant TCR Vβ population aids in the diagnosis and allows for monitoring of the disease course in SS patients. However, as SS cells express molecules also present on normal activated CD4 T cells, diagnosis based on the phenotype of circulating malignant cells can be difficult [6]. SS patients without an identifiable circulating clone pose a particular diagnostic and therapeutic monitoring challenge, as loss of CD26 is suggestive but not definitive of the malignant population [16].
To that end, much work has been performed recently to identify additional diagnostic markers for SS. Wysocka and colleagues identified CD164 as a novel early detection marker for SS in patients with low-to-high tumor burden, demonstrating a statistically significant correlation between CD164 acquisition and loss of CD26 expression [17]. The studies further showed that while CD164 is present on CD4 T cells of SS patients with a wide range of tumor burdens, it is absent on CD4 T cells of healthy controls and patients with atopic dermatitis. Additionally, morphological examination of purified CD4 + CD164+ T cells demonstrates the morphology of malignant Sézary cells. Lastly, CD4 + CD164+ T cells were noted to disappear in SS patients who experienced clinical remission as a result of treatment, underscoring the potential for CD164 to serve as a marker for malignant cells in SS.
Other markers recently reported useful in flow cytometric analysis include vimentin, CD158k [18], T-plastin (PLS3) [19], Twist [20], and NKp46 [21]. Michel and colleagues demonstrated that combination of CD158k, PLS3, and Twist gene expression profiling by quantitative PCR can be used for an efficient molecular diagnosis of SS [22]. The authors showed that CD4+ T cells from SS patients expressed significant PLS3, Twist, CD158k, and NKp46 mRNA levels and that analysis of expression of these four markers accurately classified 100 % patients in the study.
In SS, molecular testing for T-cell receptor gene rearrangements can often detect the identical clonal T-cell population in the blood and the skin. In general, matching T-cell monoclonality detectable in several different skin lesions or tissues (skin, nodes, blood) in a patient over time is suggestive of CTCL [23].
Disease Staging
Since 1979, a tumor-node-metastasis-blood (TNMB) classification and staging system has been used for MF/SS and TNMB stage has proven to be an important prognostic measure (Tables 41.2 and 41.3) [24]. In 2007, modifications were proposed by the International Society for Cutaneous Lymphomas (ISCL) regarding further stratification of blood involvement parameters to reflect the newer and more sensitive assays [25, 26]. The current B classification proposed by the ISCL includes B0 (no clinically significant blood involvement), B1 (clinically significant “minimal” blood involvement with Sézary cells <1.0 K/ul) and B2 (leukemic blood involvement detectable either as (a) Sézary cells >1.0 K/ul; (b) CD4/CD8 ratio >10 and CD4 + CD7- population >40 % or CD4 + CD26- population >30 %, (c) lymphocytosis with molecular genetic evidence of a clonal T-cell population; or (d) chromosomally abnormal T-cell clone). The updated blood involvement parameters may affect staging and prognosis of patients. Currently, the ISCL proposes that in patients with erythroderma (T4 skin classification), the B2 classification be considered the equivalent to nodal involvement (hence T4N0-1M0B2 would be upgraded from Stage III to Stage IVA1). A retrospective study by Vonderheid et al. demonstrated that using modified B criteria improved prognostication in erythrodermic patients [26]. Several large studies have since validated the 2007 ISCL staging classification for prognosis and risk stratification [27–29]. National Comprehensive Cancer Network Clinical Practice Guidelines for MF/SS have been available since 2008 and include recommendations for diagnostic and staging work up as well as current established treatments and treatment algorithms, and are updated yearly [30].
Table 41.2
TNMB classification for mycosis fungoides and Sézary syndrome, 2014 National Comprehensive Cancer Network guidelines
T (skin) | |
T1 | Limited patch/plaque (<10 % body surface area) |
T1a | Patches only |
T1b | Presence of plaques with or without patches |
T2 | Generalized patch/plaque (≥10 % body surface area) |
T2a | Patch only |
T2b | Presence of plaques with or without patches |
T3 | ≥1 tumors (≥1 cm in diameter) |
T4 | Generalized erythroderma (≥80 % BSA) |
N (lymph node) | |
N0 | No clinically abnormal (palpable; ≥1.5 cm diameter) peripheral LNs |
N1 | Clinically abnormal LNs; histopathology Dutch grade 1 or NCI LN0-2 |
N1a | Clone negative |
N1b | Clone positive |
N2 | Clinically abnormal LNs; histopathology Dutch grade 2 or NCI LN3 |
N2a | Clone negative |
N2b | Clone positive |
N3 | Clinically abnormal LNs; histopathology Dutch grade 3–4 of NCI LN4; clone positive OR negative |
M (viscera) | |
M0 | No visceral involvement |
M2 | Visceral involvement (pathology confirmation of specific organ involved) |
B (blood) | |
B0 | Absence of significant blood involvement (≤5 % of peripheral blood lymphocytes are atypical/Sézary cells) |
B0a | Clone negative |
B0b | Clone positive |
B1 | Low blood tumor burden (>5 % of peripheral `blood lymphocytes are atypical/Sézary cells but does not meet criteria of B2 |
B1a | Clone negative |
B1b | Clone positive |
B2 | High blood tumor burden defined as one of the following: ≥1000 Sézary cells/μL with positive clonal rearrangement of TCR; CD4:CD8 ratio ≥10 with positive clone; or CD4+CD7– cells ≥40 % or CD4+CD26–cells ≥30 % with positive clone |
Table 41.3
Clinical staging system for mycosis fungoides and Sézary syndrome, based on the 2014 National Comprehensive Cancer Network guidelines
Clinical stages | T | N | M | B |
|---|---|---|---|---|
IA | 1 | 0 | 0 | 0 or 1 |
IB | 2 | 0 | 0 | 0 or 1 |
IIA | 1 or 2 | 1 or 2 | 0 | 0 or 1 |
IIB | 3 | 0–2 | 0 | 0 or 1 |
IIIA | 4 | 0–2 | 0 | 0 |
IIIB | 4 | 0–2 | 0 | 1 |
IVA1 | 1–4 | 0–2 | 0 | 2 |
IVA2 | 1–4 | 3 | 0 | 0–2 |
IVB | 1–4 | 0–3 | 1 | 0–2 |
Once patients have an established diagnosis of MF/SS, a typical staging workup includes complete skin and physical examination, bloodwork including complete blood count with differential, comprehensive metabolic panel, lactate dehydrogenase, and either peripheral blood flow cytometry or Sézary cell prep (flow optional in T1a, limited patch disease with no high risk clinical or histologic characteristics). Computed tomography (CT) or positron emission tomography scanning (PET) is appropriate for patients with systemic symptoms, clinically palpable lymphadenopathy (>1.5 cm), or more advanced T classification (T1b follicular MF, extensive T2a/b, T3/tumors or T4/erythrodermic disease, patients with B symptoms). These tests are useful to assess nodal or visceral involvement by MF/SS. The role of staging bone marrow biopsy remains controversial [31]. In the United States, bone marrow biopsy is typically reserved only for patients with advanced disease with some evidence of other hematological abnormalities (i.e.: other cytopenias).
Overall survival in MF/SS is highly dependent on initial disease stage. Several studies have demonstrated that early stage disease (i.e.: skin disease only, limited to <10 % body surface area) if treated has overall median survival comparable to healthy control populations. However, with more advanced disease, overall survival also decreases with Stage IV disease having a 27 % 5-year survival and Sézary Syndrome patients having a 30 % 5-year survival [32]. In retrospective studies, the risk of disease progression ranged from 10 % (Stage IA) to 25 % (Stages IB or higher) over a 30 year period [33, 34]. While a 2003 study by Kim and colleagues [32] reported an overall survival (OS) of only 2.5 years in patients with SS, a study by Agar and colleagues [27] in 2010 reported a 3.1 year OS, and a study by Talpur and colleagues [28] from 2012 showed an OS of 4.64. As the major cause of death in patients with SS is line sepsis, which is often caused by Staphylococcus aureus, Talpur et al. report that Staphylococcus was prospectively cultured and was aggressively treated and prevented with antibiotics and skin care [35]. The authors thus postulate that the improved survival may have resulted in part from early treatment of coexisting infections, as well as earlier diagnosis of disease and advent of new therapies.
Various clinical prognostic factors have been demonstrated including patient age, initial stage, T classification, visceral disease, elevated LDH, eosinophilia, presence of Sézary cells, and B0b blood classification [27]. Important histological factors that indicate poor prognosis include “large cell transformation” (LCT, presence of >25 % large atypical T-cells in the infiltrate seen in the skin or lymph node or blood, may be either CD30+ or CD30-, with CD30- transformation having worse prognosis than CD30+) [36], folliculotropic or granulomatous infiltrate; in contrast, the presence of CD8+ tumor infiltrating cytotoxic lymphocytes (TILs) in the infiltrate has been demonstrated to be a favorable prognostic marker.
Recently, a new cutaneous lymphoma international prognostic index (CLIPi) for early and late stage CTCL has been developed and validated to aid with patient risk stratification [37]. Based on the proposed index, significant adverse prognostic factors at diagnosis included male gender, age >60, presence of plaques, folliculotropic disease, and stage N1 for early stage disease, and male gender, age >60, stages B1/B2, N2/3 and visceral involvement for late stage disease. Using these variables 3 distinct groups are designated for early and late stage patients: 0–1 (low risk), 2 (intermediate risk), and 3–5 factors (high risk). Using this prognostic index a 10 year OS for each group may be obtained (in the early stage model was 90.3 % (low), 76.2 % (intermediate) and 48.9 % (high) and 53.2 % (low), 19.8 % (intermediate) and 15.0 % (high) for the late stage model).
CLIPi thus supplements the current NCI Staging System and is a useful tool for individual prognostication and for making comparisons between different groups of patients in clinical trials.
Perhaps the most potentially powerful prognostic tool currently being developed in MF/SS is cDNA microarray analysis. Previously, MF/SS patients were shown to have a distinct gene expression profile that was distinguishable from other benign inflammatory dermatoses [38]. In addition, such profiles could also identify high risk patients with <6 month survival. The panel of genes identified included upregulated GATA3 (involved in Th2 differentiation), downregulated STAT4 (involved in Th1 differentiation), and CD26 (dipeptidylpeptidase IV, which regulates T-cell entry into the epidermis, see section “Chemokines”). Recently, several studies have confirmed that a similar panel of genes has been shown to be capable of molecularly diagnosing MF/SS patients [39, 40].
Pathophysiology
Chemokines and Their Receptors in CTCL
As mentioned previously, CTCL is a malignancy in which tumor cells exhibit an affinity for the skin. This affinity is dependent upon their expression of cell surface “trafficking” molecules which mediate their migration to the cutaneous microenvironment. Chemokines and their receptors are included in this family of molecules and play a critical role in mediating tissue-specific trafficking of leukocytes to the skin.
Chemokines are chemotactic cytokines that are produced by a wide array of tissues and range from 8 to 17 kDa in size. They are divided into families in accordance with the positioning of cysteine residues in their chemical structure. Aside from their role in cell migration, they have also been shown to play a role in the mediation of inflammatory responses, angiogenesis, and cellular proliferation [41].
Chemokine receptors are seven transmembrane-spanning G-protein coupled proteins that are capable of recognizing more than one chemokine. Several chemokines and their receptors have been identified to play a critical role in the trafficking of immune cells to the skin, more specifically, thymus and activation-regulated chemokine (TARC), macrophage-derived chemokine (MDC), stromal derived factor-1 (SDF-1), and cutaneous T-cell-attracting chemokine (CTACK). These chemokines have been shown to be produced by multiple cell types (including keratinocytes, fibroblasts, and endothelial cells) and are upregulated at sites of cutaneous inflammation. Skin-trafficking T-cells have been shown to express receptors for these chemokines.
Lymphocyte trafficking to the skin requires that the cell undergo a sequential series of events that begins with an initial tethering interaction with the endothelial surface, followed by rolling, activation, firm adhesion, and diapedesis into the dermal microenvironment. The tethering and rolling events are mediated by interactions between cutaneous lymphocyte associated antigen (CLA) and E-selectin [42]. CLA is expressed on the surface of skin-trafficking T-cells and E-selectin is expressed by endothelial cells at sites of cutaneous inflammation.
Activation of the arrest stage of lymphocyte trafficking is mediated by interactions between chemokines and their receptors. Upon engagement of the chemokine receptor, signals are transduced which activate downstream events that include a change in cellular morphology, such that integrins on the surface of the leukocyte are sufficiently exposed to contact their ligands on the dermal microvasculature (e.g. LFA-1 binding to ICAM-1). This leads to the arrest of the rolling cell, which may then migrate into the dermis along a chemotactic gradient generated by chemokines produced in the skin.
The malignant cells in CTCL have been shown to express skin-trafficking molecules and includes CLA and an array of chemokine receptors. Although frequently elevated, we and others have observed that CLA expression may be more variable among circulating malignant cells [43], whereas chemokine receptors involved in skin-trafficking are more consistently expressed at high levels among this population.
It appears somewhat counterintuitive that cells expressing skin-trafficking molecules may remain confined to the circulation, as is noted among patients with advanced disease such as in SS. This confinement of malignant cells to the blood compartment may in part be accounted for by significantly elevated chemokine levels (such as TARC) in the skin and circulation of patients. It is plausible that TARC levels in the blood may exceed a threshold concentration required for effective chemotaxis, and thus abolish a skin-derived chemotactic gradient. This idea is supported by the work of Poznansky et al. who reported the potential for T-cells to exhibit movement away from a chemotactic gradient established at concentrations above which normal directional migration is detected [44]. This finding, in combination with the variable expression levels of other skin-trafficking molecules (such as CLA) among the malignant population in CTCL may provide an explanation for the high percentage of neoplastic cells detected in the circulation of patients with advanced disease.
Several studies have evaluated the expression of chemokine receptors by malignant cells in the skin and circulation of CTCL patients. Flow cytometric analysis and immunohistochemical studies revealed preferential expression of the chemokine receptors CCR4, CXCR3 and CXCR4 among MF cells and the surrounding reactive T cells in patients with early patch and plaque stage disease. Interestingly, patients with tumor stage disease exhibit a loss of CXCR3 (and rarely CCR4), while expressing high levels of the chemokine receptor CCR7 [45]. CXCR4 and CCR4 have both been shown to play a role in lymphocyte migration to the skin, while CCR7 mediates the migration of T cells to lymphatic tissue and may account for the nodal disease observed among patients with advanced disease. CCR7 recognizes secondary lymphoid tissue chemokine (SLC/CCL21) produced within the nodal microenvironment. Preferential expression of CCR10 has also been reported among tumor cells of MF patients. CCR10 is the receptor for the cutaneous T-cell attracting chemokine (CTACK/CCL27) which is constitutively produced by epidermal keratinocytes [46].
Similar to the MF cells in tumor stage disease, the malignant cells in patients with peripheral blood disease have been shown to express elevated levels of CCR4, CCR7, CXCR4, and CCR10 [47]. In addition, the circulating malignant cells are also characterized by their loss of the cluster of differentiation markers CD7 and/or CD26 [48]. The absence of CD26 is believed to contribute to the accumulation of malignant cells in the skin of Sézary syndrome patients [49].
Campbell and colleagues have recently demonstrated that MF and SS may arise from two distinct T-cell subsets. Their studies show that clonal malignant T cells from the blood of SS patients universally coexpress the lymph node homing molecules CCR7 and L-selectin as well as the differentiation marker CD27, a phenotype consistent with central memory T cells (TCM) [50]. The authors also note that CCR4 was universally expressed at high levels, while there was variable expression of CCR6, CCR10, and CLA. In contrast, T cells isolated from MF skin lesions lacked CCR7/L-selectin and CD27, but strongly expressed CCR4 and CLA, a phenotype of skin resident effector memory T cells (TEM).
Additionally, genomic analysis reveals differing molecular profiles characteristic of MF and SS. MF is characterized by increased expression of FASTK and SKAP1 genes, and diminished expression of the RB1 and DLEU1 tumor suppressor genes [51], while SS is characterized by amplification of MYC oncogene in 75 % of patients with SS, which was detected in only a minority of patients with MF [52].
Although MF and SS were originally thought to represent points of progression in a disease continuum, the recent data suggest that these two entities might best be considered as separate lymphoproliferative diseases which originate from distinct T-cell subsets and have different genetic signatures.
CD26 is a dipeptidyl transferase expressed as both a soluble factor in the circulation and as a cell surface protein expressed by lymphocytes. CD26 mediates the cleavage of the chemokine stromal derived factor-1 (SDF-1), produced by inflamed skin (Fig. 41.2a). SDF-1 is the natural ligand for the chemokine receptor CXCR4, which is expressed by the malignant population in Sézary syndrome. The absence of CD26 leads to elevated levels of SDF-1, which in turn, contributes to the trafficking of malignant cells to skin (see Fig. 41.2b).
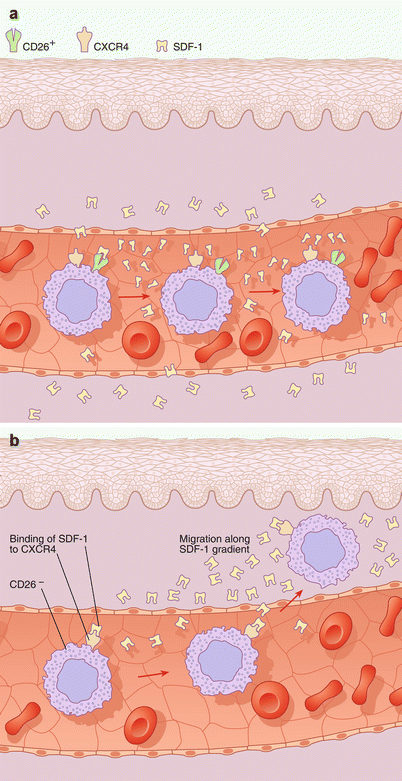
Fig. 41.2
(a) CD26 is a bipeptidyl transferase expressed by normal T lymphocytes that induces the cleavage and inactivation of the chemokine stromal-derived factor-1 (SDF-1). This cleavage prevents the binding of SDF-1 to its receptor, CXCR4, thus interfering with SDF-1/CXCR4-mediated chemotaxis into the skin. CD26+ T cells are resistant to the chemotactic effects of SDF-1 via CD26-mediated inactivation (top). (b) The absence of CD26 is reported among circulating malignant cells in Sézary syndrome. In the absence of CD26, SDF-1 may efficiently establish a chemotactic gradient that promotes the migration of malignant cells into the skin via engagement of CXCR4. Engagement of CXCR4 by SDF-1 in the cutaneous microvasculature and subsequent chemotaxis of the cell into the dermal microenvironment are demonstrated (top)
A CD26 negative population of greater than 30 % among erythrodermic patients is believed to be sufficient to support a diagnosis of Sézary syndrome, and thus differentiate such patients from individuals suffering from benign inflammatory dermatoses [16]. We have recently reported a correlation between the percentage of circulating CD4 + CD26- T-cells and changes in clinical status among Sézary syndrome patients. More specifically, a reduction in this population typically heralds improved clinical status, whereas, an increase frequently precedes disease progression characterized by a greater than 20 % increase in affected body surface area, or a greater than 10 % increase in body surface area accompanied by progressive lymphadenopathy and/or hepatosplenomegaly [53].
While chemokine receptors are necessary for tissue-specific lymphocyte trafficking to the skin, expression of their chemokine ligands plays an essential role in this process. In the absence of chemokines, engagement and activation of the chemokine receptor will not occur. The epidermotropism exhibited by neoplastic cells in CTCL has been attributed, in part, to the production of chemokines by epidermal cells. Whether their production arises secondary to the accumulation of malignant cells in the skin or precedes the infiltration has not be en established.
Elevated levels of the chemokine, cutaneous T-cell attracting chemokine (CTACK/CCL27), has recently been reported in the serum of patients with CTCL. The highest levels were detected among patients with more advanced disease (tumor stage and erythroderma), and improved in response to successful treatment [54]. CTACK has been shown to be produced by epidermal keratinocytes and is presented on the surface of dermal endothelial cells [46, 55]. It is recognized by the chemokine receptor, CCR10, which, as mentioned previously, is expressed by the malignant population [56].
Thymus and activation-regulated chemokine (TARC/CCL17), the ligand for CCR4, was shown to be elevated in CTCL patients relative to normal individuals and patients with psoriasis vulgaris [57]. More specifically, an increase in serum TARC levels was shown to correlate with more advanced disease, being greatest among tumor stage patients as compared to patch and plaque stage patients. Immunohistochemistry revealed TARC expression by lesional keratinocytes at all stages of disease. These findings support a functional role for CCR4 in the accumulation of malignant cells in the skin of CTCL patients.
Preferential expression of mRNA for the chemokine interferon-gamma inducible protein 10 (IP-10) has been reported in the epidermis of patients with early stage epidermotropic CTCL [58]. IP-10 is the ligand for CXCR3, which is expressed by MF cells in early patch/plaque stage disease [45]. CXCR3 and its ligand, IP-10, are considered to be Th1 associated molecules given their ability to recruit Th1 cells to sites of inflammation. Thus, levels of IP-10 and CXCR3 positive cells may reflect the host anti-tumor response. Patients with more advanced disease exhibit a depression in CXCR3 levels, whereas, expression of the Th2 associated chemokine-receptor, CCR4, and its ligand (TARC), remain elevated and skew the cutaneous cytokine milieu in favor of Th2 cytokine production, which likely contributes towards the pathogenesis of disease in CTCL.
Once the malignant T-cells have been recruited to the skin, their growth appears to be supported by the in situ production of cutaneous cytokines. Yamanaka and colleagues have demonstrated that lesional skin of CTCL patients over produces interleukin-7 [59]. IL-7 itself clearly exhibits pro-proliferative effects on malignant T-cells derived from CTCL patients.
Chromosomal and Genetic Abnormalities
MF/SS is not considered a primary genetic disorder attributable to discrete mutation(s), however with disease progression, several genetic abnormalities have been detected. Cytogenetic studies have demonstrated chromosomal deletions (1p, 17p, 10q, 19) and gains (4q, 18, 17q) [60]. Even early MF lesions have been shown to have STAT 3 constitutively activated or defects in their apoptosis pathways (decreased Fas expression) [61–63]. In more advanced disease, such as tumor-stage MF or SS, other genetic perturbations include p15/16 and p53 defects, microsatellite instability, hypermethylation of tumor suppressor genes (p14, p15, p16, BCL7a, PTPRG, p73) [64, 65], and constitutive activation of NF-kB [66]. In addition to these, epigenetic factors (which affect gene expression through modification of histones and other chromatin-associated proteins) appear to play a role in CTCL development/progression, given the clinical activity of histone deacetylase inhibitors (HDACi) in CTCL [67].
MF/SS malignant T-cells also can variably express the interleukin-2 receptor (IL-2R) that binds the cytokine IL-2. IL-2R is comprised of 3 subunits: IL-2Rα (CD25/p55), IL-2Rβ (CD122/p75) and IL-2Rγ (CD132/p64). IL-2Rα has been viewed as a marker of cell activation and is significantly expressed in approximately 20 % of MF/SS patients, particularly in later stage disease [68], and soluble IL-2R can be detected in the peripheral blood [69]. The T-cells can also express other activation markers such as CD45RO and/or proliferating-cell nuclear antigen (PCNA).
Immunobiology
Immune Dysregulation in CTCL: Clues to Understanding Disease Progression
It has long been known that patients with advanced MF/SS typically exhibit abnormalities in a variety of parameters of cell-mediated immunity. Moreover, the malignant T-cells have been implicated as the cause of the endogenous immune deficiency. Earlier observations by Vowels and colleagues demonstrated that the malignant CD4+ T cells observed in most cases of MF/SS appear to exhibit a Th2 phenotype (Fig. 41.3). In vitro stimulation of peripheral blood cells derived from SS patients routinely results in increased levels of measurable IL-4 expression [70]. Furthermore, Vowels et al. demonstrated levels of IL-4 and IL-5 mRNA in clinically involved skin, even among patients with early patches or plaques while uninvolved skin and the skin of normal volunteers did not have detectable levels of Th2 cytokine mRNA [71]. Increasing levels of IL-10 mRNA in parallel with an increasing density of the malignant T cell infiltrate as lesions progressed from patch to plaque to tumor has also been demonstrated [72]. cDNA microarray analysis of the malignant T-cells isolated from patients with SS have shown that the Th2 cell–specific transcription factors such as GATA-3 and Jun B are highly overexpressed [38]. Thus, despite evidence of a vigorous host response in skin lesions in early disease, characterized by the presence of IFN-γ–secreting CD8+/TiA-1+ T cells [73], the chronic production of Th2 cytokines such IL-4, IL-5 and IL-10 by the malignant T cell population likely represents one mechanism by which the tumor cells circumvent the anti-tumor immune response.
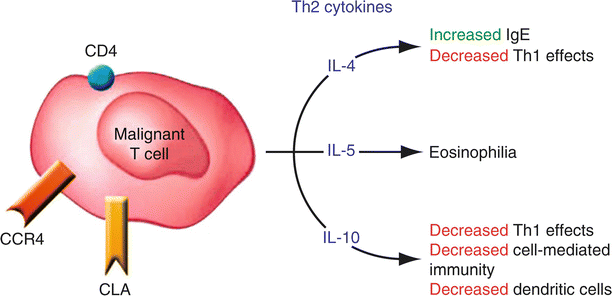
Fig. 41.3
The malignant T cell in MF/SS expresses CD4/CLA/CCR4/CCR10 and produces the cytokines IL-4/IL-5/IL-10, which results in Th2 predominance and subsequent multiple abnormalities in cellular immunity (From Kim et al. [5] with permission of the American Society for Clinical Investigation. Copyright 2005)
The host anti-tumor response, although effete, may play a role, albeit small, in containment of disease progression, even among those with advanced disease. This is supported by the observation of rapid progression of SS associated with the use of immunosuppressive agents such as cyclosporine [74]. In addition to enhanced Th2 cytokine production during disease progression, patients with circulating malignant T cells also manifest defects in Th1 cytokine production. Wysocka et al. observed a progressive decline in the production of IL-12 and IFN-α by peripheral blood cells that correlated with an increase in the peripheral blood burden of malignant T cells [75]. Furthermore, the decline in production of these cytokines appeared to be linked to a decline in the numbers of peripheral blood myeloid and plasmacytoid DCs, respectively. Accompanying the decrease in DCs and a defect in IL-12 production is a deficit in production of other products of myeloid DCs, including IL-15 which is an important IFN−γ–stimulating agent and powerful booster of Th1 responses. In contrast,Yamanaka et al., observed increased serum IL-18 and IL-18 mRNA within CTCL skin samples suggesting alternative potential sources for this interferon inducing cytokine [76] Nevertheless, peripheral blood cells of SS patients clearly manifest marked decreases in IFN-γ production [70].
Intense recent interest has focused on the possible contributory role of regulatory T-cells (Tregs) in the endogenous immunosuppression in advanced CTCL. Berger and colleagues have demonstrated that under certain in vitro conditions that malignant T-cells may assume properties of Tregs, including the expression of Foxp3 and production of TGF-β and IL-10 [77]. Similarly, Walsh et al., was able to demonstrate increased Foxp3 expression among the peripheral blood cells of some patients with SS and among the circulating cells of all patients with HTLV-1 associated CTCL [78]. The cells of such patients were found to produce increased concentrations of TGF-β. Wong et al. has observed increased expression of CTLA-4 upon activation of the circulating cells of patients with CTCL, indicating that this molecule, which is typically associated with Tregs, was upregulated and a sign of increased numbers of Treg cells [79]. These findings remain controversial as Tiemessen observed that the Foxp3 positive Tregs of some CTCL patients are dysfunctional and exhibit a reduced ability to inhibit the proliferation of CD4+/CD25- T-cells [80] while Yamano and colleagues have made similar observations in regard to HTLV-1 infected CD4+ cells [81]. Despite these disparate findings, many investigators involved in studies of the immunobiology of CTCL feel that Tregs likely play some role in the pathogenesis of the immune suppression. Thus, as discussed below, elimination of Tregs is just one of several new therapeutic strategies under investigation.
Other mechanisms that could account for abnormalities in DC maturation, and thus, for diminished IL-12 production have been highlighted by recent studies by French et al., which demonstrated a defect in expression of CD40 ligand upon activation of malignant T cells derived from patients with SS [82]. CD40 ligand is not expressed on resting T cells but is normally upregulated on the cell surface upon engagement of the TCR. By contrast, malignant CD+/CD7- T cells fail to express CD40 ligand upon engagement of the TCR by anti-CD3. Clearly, the absence of CD40 ligand interaction with CD40 on APCs during an immune response can lead to a profound reduction in DC activation and cytokine production. Through the in vitro addition of recombinant hexameric CD40 ligand, French and colleagues demonstrated reconstitution of IL-12 and TNF production by the cells of patients with SS. These findings provide obvious insights regarding potential strategies for correcting defects in immune dysregulation related to diminished numbers and function of DCs associated with advancing MF/SS.
Derangements in T-cell diversity may also play a role in the immune suppression of CTCL. Yawalkar et al., using beta-variable complementarity-determining region 3 spectratyping determined that patients with both early as well as late disease exhibit loss of the normal T-cell repertoire [83, 84]. These findings are reminiscent of a chronic retroviral infection as similar observations have been made in HIV infection. Loss of T-cell repertoire may partially account for the failure of advanced stage CTCL patients to respond normally to a variety of antigens.
The result of the diverse abnormalities of DC and T-cell function during progressive MF/SS are multiple abnormalities in cellular immunity (Fig. 41.4). Marked defects in cellular cytotoxicity occur that correlate with the burden of circulating malignant T cells [85]. Progression from early to more advanced MF/SS is typically associated with a marked decline in NK cell numbers and activity which could partially be due to a decline in myeloid DC production of IL-15 which is critical for the normal growth of these cells. Similarly, a decline in the number of peripheral blood CD8+ T cells accompanies an increasing burden of circulating malignant T cells. Furthermore, the percentage of these cytotoxic cells that express activation markers, such as CD69, is also significantly reduced compared to NK and CD8+ T cells from MF patients without overt peripheral blood involvement [86]. A reduction in the number of functioning NK and CD8+ T cells is almost certainly associated with a deterioration of both host anti-tumor immunity and immune surveillance against microbial organisms. Examples of these phenomena include infections such as disseminated herpes simplex/zoster or progressive multifocal leukoencephalopathy among SS patients who have never been iatrogenically immunosuppressed by chemotherapy or other immunosuppressive medications [8, 87, 88]. As mentioned earlier, other malignant neoplasms, including melanoma and non-melanoma skin cancers as well as Hodgkin’s lymphoma, also appear to be more common in MF/SS patients independent of the history of previous predisposing therapy (such as phototherapy or radiation therapy) [10, 89, 90]. These findings reflect an overall impairment in immune surveillance against cancer.
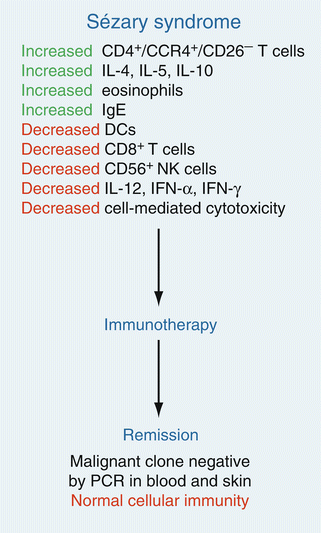
Fig. 41.4
Elimination of the malignant T-cell clone during immunotherapy leads to a restoration of a normal immune response. Studies of numerous patients with SS have demonstrated that induction of complete remission with clearing of the malignant T-cell clone during multimodality immunotherapy leads to a restoration of normal host immune function (From Kim et al. [5] with permission of the American Society for Clinical Investigation. Copyright 2005)
Other characteristic immunological findings associated with the progression of MF/SS include development of peripheral eosinophilia and elevated levels of serum IgE [70, 91]. Peripheral eosinophilia has been determined to be an independent marker for poor prognosis and disease progression [92]. In one study, peripheral blood cells from patients with SS and eosinophilia produced markedly higher levels of IL-5 upon stimulation than did cells of patients or normal volunteers without eosinophilia [93]. A finding of importance in this study indicated that culture of the patients’ cells with either recombinant IFN-α or IL-12 significantly inhibited the excess production of IL-5. These observations suggest that these cytokines could be useful therapeutic tools to prevent continued proliferation of eosinophils under the influence of IL-5, thus, possibly preventing at least some of the adverse effects associated with high eosinophil counts.
It is particularly noteworthy that after successful treatment of SS with immunotherapeutic agents, with resolution of evidence for skin disease and disappearance of the malignant clone from the blood, virtually all abnormal immune parameters are restored to normalcy (Fig. 41.4) [85]. One implication of these observations is that the malignant clone is likely responsible for much of the immune dysregulation that occurs in SS. Moreover, these findings indicate that SS patients who experience remission will have their immune system at least partially reconstituted. Thus, such patients should be less likely to experience severe consequences of microbial infection in comparison to those with advanced SS.
Treatment
Treatment of MF/SS can be divided into 2 major categories: skin directed therapies (SDTs) and systemic therapies. As listed in Table 41.4, there are numerous options. Therapeutic approaches can vary widely from center to center, and the availability of many newer therapies varies among countries. It is useful to divide the discussion of therapies in regard to early stage vs. late stage disease (Table 41.5).
Table 41.4
Treatment options in MF/SS
Skin directed therapy | Mechanism of action |
Topical corticosteroids | Tumor cell apoptosis, decrease skin LCs |
Topical chemotherapy (nitrogen mustard, BCNU) | Tumor cell apoptosis |
Topical retinoids (bexarotene, tazarotene) | Tumor cell apoptosis |
Topical imiquimod | TLR7 agonist, triggers innate & adaptive anti-tumor immunity |
Phototherapy (UVB, PUVA, excimer laser) | Tumor cell apoptosis Decrease skin LCs |
Electron beam therapy (EBT) | Tumor cell apoptosis |
Biological therapy | |
RXR retinoid (bexarotene) | Tumor cell apoptosis Inhibit tumor cell IL-4 production |
RAR retinoid (isotretinoin, all-trans retinoic acid) | Tumor cell apoptosis Induce interferon-γ |
Interferons (alpha, gamma) | Enhanced cell-mediated cytotoxicity Inhibit tumor cell Th2 cytokine production |
GM-CSF | Enhanced circulating dendritic cell numbers and function |
Extracorporeal photopheresis | Circulating tumor cell apoptosis Induction of DC differentiation |
Fusion protein/toxin (denileukin diftitoxa) | Targets and kills CD25 (IL2 receptor) expressing tumor cells. |
Histone deacetylase inhibitors (vorinostat, romidepsin) | Affects tumor cell gene transcription, non-histone effects. |
Other systemic therapy | |
Cytotoxic chemotherapy (MTX, pegylated doxorubicin, gemcitabine, etoposide, pentostatin) | Cytotoxic agents |
Hematopoeitic stem cell transplantation (allogeneic) | Cytotoxic agents (induction) |
Graft-vs-tumor effect | |
Experimental therapy | |
Transimmunization ECP | Enhances DCs antigen processing of apoptotic tumor cells |
Targeted monoclonal antibodies (CD4, CD52, CD40, CCR4) | Targets tumor cells (CD4, CD52, CCR4) Activated dendritic cells (CD40) |
Cytokines (IL12, IL2, IL15) | Augments cell mediated anti-tumor immunity |
Toll-like receptor agonists (CpG-ODN, imidazoquinolones) | TLR agonists, augment innate and adaptive anti-tumor immunity |
Tumor vaccines | Clonotypic TCR as antigen Dendritic cell based vaccines |
Table 41.5
Treatment of MF/SS by clinical stage
Stage | Initial therapy | Subsequent therapy | Therapy for refractory disease |
|---|---|---|---|
IA (early stage) | Skin directed therapies | ||
IB/IIA | Topical chemotherapy Phototherapy Electron beam therapy (± low dose biological agents) | Interferons Retinoids Multimodality rx Topical chemo + biological agent Phototherapy + biological agent 2 biological agents Denileukin diftitoxa Histone deacetylase inhibitors | Experimental therapies |
IIB (advanced stage) | Few tumors Localized EBT Intralesional IFN Topical chemotherapy + biological agent Generalized tumors Total skin EBT Denileukin diftitox Multimodality therapies | Multimodality therapy Denileukin diftitox Histone deacetylase inhibitors Single agent chemotherapy | Experimental therapies |
IIIA, B | PUVA Retinoids Interferons Methotrexate ECP Histone deacetylase inhibitors Multimodality therapies | Multimodality therapies Denileukin diftitox Histone deacetylase inhibitors Single agent chemotherapy | Experimental therapies |
IVA, B | Single agent systemic therapy Multimodality therapy (+skin directed therapy) | Adjuvant palliative local radiation for extracutaneous disease Bone Marrow/Stem Cell Transplant Chemotherapy | Experimental therapies |
Early MF
It has become quite clear during the past several decades that traditional systemic chemotherapy has not resulted in durable remissions in MF/SS [1]. As a consequence, emerging therapeutic efforts have focused upon targeted biological agents and immune modifications using a multimodality approach. Numerous arms of the immune system must cooperate to generate a sufficient host anti-tumor response such that the proliferation of the malignant T cell population in MF/SS patients can be controlled and, ideally, eradicated (Fig. 41.5).
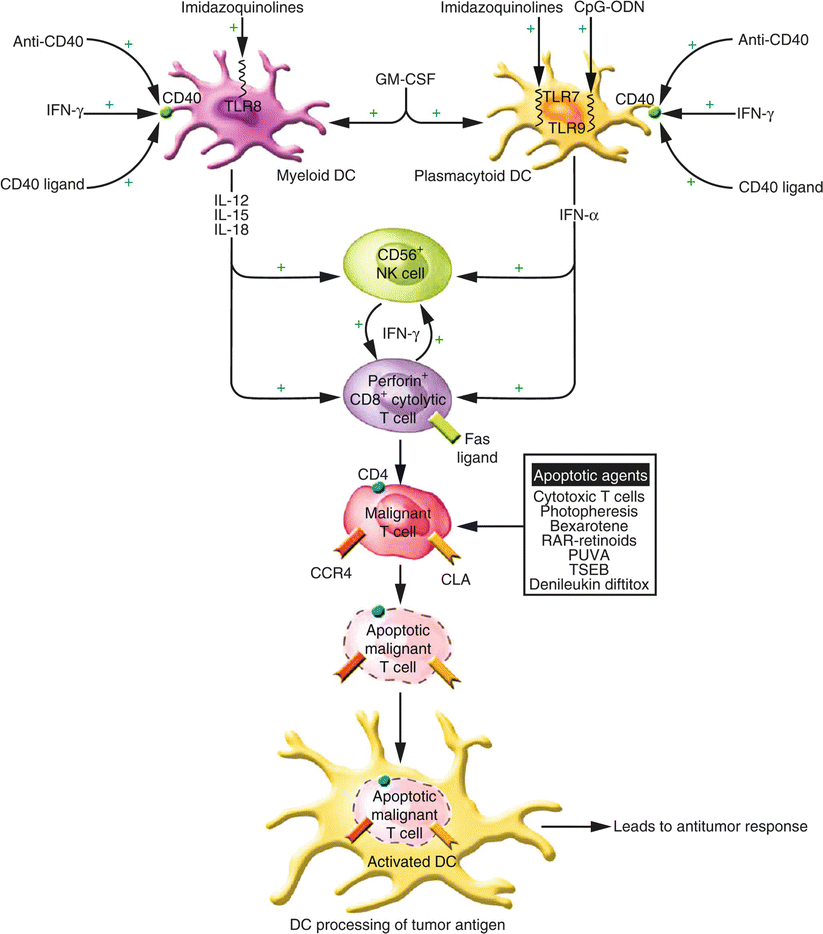
Fig. 41.5
Multimodality strategy for enhancing the antitumor response using immunotherapeutics. A multimodality approach encompasses the activation of multiple arms of the immune response through the use of agents to activate DCs, CTLs (CD8+), and NK cells (CD56+). Granulocyte-macrophage colony-stimulating factor (GM-CSF) may enhance the numbers of DCs while agents that enhance CD40 expression (IFN-γ) and activation (CD40 ligand; activating anti-CD40 antibody) and TLR ligands (CpG-ODNs; imidazoquinolines) lead to DC cytokine production and to enhanced DC processing of apoptotic malignant T cells. Cytokines produced by DCs as well as exogenously administered cytokines augment CD8+ T-cell cytokine production. Proapoptotic agents, including bexarotene, RARspecific retinoids, PUVA, photopheresis, topical chemotherapy, total skin electron beam irradiation (TSEB), and denileukin diftitox can assist in the development of an antitumor immune response by reducing the overall tumor burden and by providing a source of apoptotic malignant cells and tumor antigens for uptake by DCs. (From Kim et al. [5] with permission of the American Society for Clinical Investigation. Copyright 2005)
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


