Neuromediator
Source
Receptor
Primary function
Substance P
Sensory nerve fibers
Meissner corpuscles
Perivascular nerves
Mast cells
Monocytes
Eosinophils
Tachykinin (neurokinin) receptor
Mediates erythema and edema
Increases histamine and TNF-α release from mast cells
Upregulates cell adhesion molecule expression in dermal endothelial cells
Stimulates release of proinflammatory cytokines from keratinocytes
VIP
Sensory nerve fibers
Sweat glands
Merkel cells
PMNs
VPAC1 and VPAC2 receptors
Vasodilation
Increases sweat secretion
Histamine release from mast cells
Keratinocyte proliferation and migration
Inhibits Langerhans cell antigen presentation for Th1 responses while augmenting Th2 and Th17 responses
PACAP
Sensory nerve fibers
Autonomic nerves
Lymphocytes
Endothelial cells
VPAC1 and VPAC2 receptors
Vasodilation
Down-regulates proinflammatory cytokines in T cells
Modulates mast cells
Inhibits NF-κB
Inhibits Langerhans cell antigen presentation for Th1 responses while augmenting Th2 and Th17 responses
CGRP
Sensory nerve fibers
Perivascular nerves
Meissner corpuscles
CGRP receptors
Vasodilation
Keratinocyte proliferation
Histamine release (mast cells)
Inhibits NF-κB
Inhibits Langerhans cell antigen presentation for Th1 responses while augmenting Th2 responses
POMC peptides
Keratinocytes
Melanocytes
Langerhans cells
Fibroblasts
Mast cells
Monocytes
Macrophages
Endothelial cells
PMNs
Sensory nerves
Melanocortin receptors
Immunomodulation (upregulates IL-10 and antagonizes effects of proinflammatory cytokines)
Inhibits NF-κB
Melanogenesis
Catecholamines
Autonomic adrenergic nerves
Keratinocytes
Melanocytes
Adrenergic receptors
Inhibit keratinocyte migration
Regulates natural killer and monocyte activity
Inhibits Langerhans cell antigen presentation for Th1 responses
NPY
Perivascular nerves
Langerhans cells
G protein-coupled Y1-Y5 rhodopsin-type receptors
Vasoconstriction
Eccrine sweat production
Antimicrobial properties
ATP
Numerous sources with intracellular and/or extracellular activity
Keratinocytes
Sympathetic nerve fibers
Ligand-gated purinergic P2X receptors
G protein-coupled purinergic P2Y receptors
Regulation of cell growth and differentiation
Immunomodulation [promotes dermal microvascular endothelial cell production of IL-6, GRO-α (CXCL1) and IL-8 (CXCL8), MCP-1 (CCL2) production; IL-1 release by macrophages]
Enhance Langerhans cell antigen presentation
Catestatin
Sensory nerve fibers
Adrenal chromaffin cells
Adrenergic nerve fibers
Immune cells
Nicotinic acetylcholine receptors
G protein-coupled receptors
Keratinocyte migration
Stimulation of monocyte and PMN chemotaxis
Mast cell degranulation
Pro-inflammatory cyto- and chemokine production
Antimicrobial properties
The distribution of NPs varies depending on the body site. High levels of SP, NKA and CGRP are found in areas with the greatest tactile sensation. Intermediate levels are found in the neck and face, whereas the lowest levels are present in the groin, arm and thigh [16]. Levels of VIP and peptide histidine methionine (PHM) are also highest in axillary skin, suggesting their role in axillary eccrine sweat production. The location of NPs also varies based upon the layer of the skin in question. SP, NKA and CGRP are found in nerves penetrating the epidermis and the neuropeptides SP, CGRP, VIP and NKA are in nerves innervating dermal structures [9]. NPs bind to specific receptors on nerves and other cell types and, in turn, activate intracellular signaling cascades. NPs are then inactivated by peptidases such as neutral endopeptidase or angiotensin-converting enzyme (ACE) [17, 18].
Delineating the myriad functions of neuropeptides and neurohormones has led to the emergence and explosive growth of a field investigating the intimate relationship between the nervous, immune and endocrine systems and the skin. Terms such as the neuro-immune-cutaneous system are used to imply a relationship between immune and non-immune cells in the skin and between the immune system and the nervous system within the skin [19].
The skin is an ideal organ in which to investigate these relationships due to its location, size and function. As the interface between the external and internal environments the skin has a unique role in modulating and transmitting outside stimuli while maintaining internal homeostasis. As a result, there is increasing attention and focus on the skin as a site for unraveling these connections, applying them to other organ systems and ultimately, using them as a model for therapeutic interventions in several disease states. In this chapter we highlight several NPs and other neural signals, discuss their functions and describe their role in clinical skin disorders (Table 12.1).
Receptors
There are numerous neuropeptide receptors. For example, SP has three receptors, NK1, NK2, NK3. Two receptors have been identified for CGRP (1,2) and NPY has receptors referred to as Y1,2,3,4,5 [20]. Most cutaneous cells express several receptor types; to discuss each in detail is beyond the scope of this chapter, and more comprehensive reviews are available [9]. Rather, the receptors most relevant to the skin will be discussed. These include NK1, preferentially expressed on human keratinocytes [2], VPAC-1R (vasoactive intestinal polypeptide/pituitary adenylate cyclase activating polypeptide 1 receptor) on dermal endothelial cells and Langerhans cells [21], VPAC-2R on keratinocytes and Langerhans cells [21] and melanocortin 1 receptor (MC-1R) on melanocytes, keratinocytes, monocytes, fibroblasts and Langerhans cells [9]. Additionally, CGRP has effects on many cellular components of skin that can be blocked by competitive inhibitors of the type I CGRP receptor (see below) suggesting the presence of CGRP receptors on various cell types within the skin.
Substance P (SP)
SP is the best-characterized neuropeptide. An undecapeptide, it is a member of the tachykinin family along with NKA and neurokinin B (NKB). SP is released from sensory nerves that are in contact with endothelial cells, mast cells, hair follicles and epidermal cells [22]. Its biological effects in the skin are mediated predominantly by the NK1 receptor. SP receptor-binding leads to activation of phospholipase C, an increase in intracellular calcium and subsequent activation of nuclear factor-kappa B (NF-kB) [23].
SP is a potent vasodilator, accounting for its role in the wheal and flare response in neurogenic inflammation. SP directly acts on vascular smooth muscle and indirectly on the endothelium to enhance the production of nitric oxide, which contributes to vasodilatation and increased vascular permeability [24]. In human skin, SP is also released by free nerve endings in the dermal papilla and epidermis of human fingers, in Meissner corpuscles, and near sweat gland ducts and blood vessels [12]. Receptors for SP are found on mast cells, lymphocytes, leukocytes and macrophages. SP-stimulated macrophages generate prostaglandin E2, thromboxane B2 and superoxide ion, and SP-stimulated keratinocytes release proinflammatory cytokines IL-1α, IL-1β and IL-8 [2]. Furthermore, SP induces histamine release from mast cells, lymphocyte proliferation and chemotaxis, immunoglobulin production and the release of cytokines IL-1, IL-6 and TNF-α [25]. An increase in SP-expressing nerve fibers is seen in inflammatory skin diseases such as atopic dermatitis [26]. SP increases the release of proinflammatory cytokines from keratinocytes and TNF-α, histamine, leukotriene B4 and prostaglandin D2 from mast cells. SP also upregulates IL-2 production to promote T-cell proliferation and induces the expression of the adhesion molecules P-selectin, intercellular adhesion molecule (ICAM) 1 and vascular cell adhesion molecule (VCAM) 1 [2, 26]. Recent evidence suggests that SP also plays a role in regulating cutaneous microflora [27]. SP-treated bacteria populations including B. cereus were found to have increased biofilm formation activity. Furthermore, SP enhanced B. cereus-induced cytotoxic activity against keratinocytes, evidenced by increased caspase 1 production and morphological changes characteristic of necrosis [27]. The effect of SP on the cutaneous microbiome may influence a variety of skin disorders [27].
Pituitary Adenylate Cyclase Activating Polypeptide (PACAP) and Vasoactive Intestinal Peptide (VIP)
PACAP exists in two forms, PACAP-38 and a truncated form, PACAP-27 [28]. PACAP belongs to the glucagon-secretin family that includes VIP and growth hormone-releasing hormone. PACAP binds to two types of receptors. PAC1 binds PACAP to activate adenylate cyclase and phospholipase C, whereas a second type of receptors, VPAC1 and VPAC2, bind both PACAP and VIP to activate adenylate cyclase [14]. PACAP has the highest immunoreactivity around blood vessels, hair follicles and sweat glands, and has been shown to modulate inflammatory responses in the skin [2].
VIP is a 28-amino-acid peptide that is localized in the deeper dermis in nerve fibers situated around eccrine sweat glands, superficial and deep vascular plexuses and hair follicles [24]. VIP is a potent vasodilator that contributes to the development of pruritus, erythema and edema. VIP also plays a role in the regulation of cutaneous blood flow, the promotion of nitric oxide synthesis, keratinocyte proliferation and sweat production. VPAC-1R (vasoactive intestinal polypeptide/pituitary adenylate cyclase activating polypeptide 1 receptor) is the dominant receptor on human dermal endothelial cells, whereas VPAC-2R is expressed on keratinocytes [2]. Both receptor types are found on Langerhans cells [21].
In a murine model, intradermal administration of PACAP suppressed the induction of contact hypersensitivity (CHS) at the injected site and in vitro treatment of epidermal antigen presenting cells inhibits their ability to present antigen for elicitation of delayed-type hypersensitivity (DTH) in previously immunized mice [14]. PACAP also inhibited the ability of Langerhans cells to present antigen in wholly in vitro assay systems for a Th1 response [14] . In vitro studies suggest this may be due to PACAP-induced suppression of IL-1β release and augmentation of IL-10 production [14]. In a mouse model, VIP also inhibited the ability of epidermal cells enriched for Langerhans cell content to present antigen for elicitation of delayed-type hypersensitivity in previously immunized mice [13]. Furthermore, VIP inhibited Langerhans cell antigen presenting capability in in vitro assays of antigen presentation for Th1 responses [13].
The inhibitory effects of PACAP, VIP and CGRP on antigen presentation may occur through suppression of NF-kB, which is known to be involved in regulating transcription of the TNF-α gene [29], as well as CD86 and other pro-inflammatory cytokines [14].
While VIP and PACAP inhibit Langerhans cell antigen presentation for Th1 immune responses, these NPs enhance antigen presentation to generate Th2 cells [30]. In vivo promotion of Th2 responses by VIP and PACAP signaling has also been demonstrated through manipulation of VPAC2 expression in a transgenic murine model; overexpression of VPAC2 in CD4+ T cells biased toward Th2 immunity, whereas mice deficient in VPAC2 demonstrated predominance of the Th1 response [31].
Th17 cells are a subset of CD4+ T cells that produce a variety of cytokines of the IL-17 family, as well as IL-21 and IL-22. IL-21 production may lead to further differentiation of more Th17 cells, while IL-22 plays a role in keratinocyte proliferation and epidermal hyperplasia [32]. Some Th17 cells produce IL-17A but only very little IL-22 [33].
Th22 cells are CD4+ T cells that produce IL-22 but not IL-17A. IL-22 is known to act on epithelial cells such as keratinocytes to trigger antimicrobial peptide production [30]. VIP and PACAP have been shown in vitro to bias Langerhans cell antigen presentation toward Th17, in addition to Th2 immune responses. Exposure of Langerhans cells to PACAP or VIP followed by antigen presentation to CD4+ T cells led to enhanced production of IL-17A and IL-4 by CD4+ T cells with suppressed production of IL-22 and IFN-γ [30]. Furthermore, after antigen presentation the CD4+ T cell population had enhanced number of cells expressing intracellular IL-17A or IL-4 with decreased numbers of cells expressing IFNγ [30]. In accordance with these findings, the Th17 cell-associated transcription factor RORγT and the Th2-associated transcription factor GATA3 were elevated while T-bet (Th1-associated) was decreased [30].
In vivo studies in which PACAP was applied topically to human skin revealed that PACAP induces marked edema in a concentration-dependent manner [34]. Investigators also injected PACAP intravenously and observed a concentration-dependent vascular response (flushing, erythema and edema) as well as a 2.7 °C rise in body temperature [34]. Together, this evidence suggests a role for PACAP in the vasoactive component of neurogenic inflammation found in skin disorders such as rosacea, urticaria and atopic dermatitis [34].
Recent experiments demonstrated that human keratinocytes significantly upregulated vascular endothelial growth factor (VEGF) production in response to VIP stimulation in a dose- and time-dependent manner [35]. Maximum enhancement of VEGF production was observed with the combination of VIP and IFN-γ stimulation [35]. This mechanism might underlie the angiogenesis and vasodilatation characteristic of lesional psoriasis skin [35].
Calcitonin Gene Related Peptide (CGRP)
CGRP is a 37-amino-acid neuropeptide discovered in 1982 [36]. There are two forms, CGRP-α, or CGRP-1, and CGRP-β, or CGRP-2. In human skin, CGRP is co-localized with SP in nerves in the dermal papillae and free nerve endings of glabrous skin; however, when co-localized with somatostatin, it is found in nerve fibers associated with the epidermis and perivascular space [24]. CGRP is among the most prevalent cutaneous NPs and is found in association with mast cells, melanocytes, keratinocytes, Langerhans cells and Merkel cells [2, 22].
CGRP inhibits antigen presentation by macrophages [37] and blood-derived dendritic cells [38, 39] and upregulates melanocyte proliferation, dendricity and melanogenesis [19]. Early observations of CGRP-containing unmyelinated nerve fibers in direct contact with the surface of epidermal Langerhans cells suggested an intimate relationship [40] (Fig. 12.1). While CGRP inhibits Langerhans cell antigen presentation in Th1 responses [40], it enhances Langerhans cell antigen presentation for Th2 responses, as evidenced by increased IL-4 production [41]. It also stimulates Langerhans cell production of the Th2 chemokines CXCL17 and CXCL22 while inhibiting the stimulated production of the Th1 chemokines CXCL9 and CXCL10 [40]. CGRP effects on LC antigen presentation for Th1 responses can be inhibited by CGRP8–37, a competitive inhibitor of the type I CGRP receptor [42]. Recent in vivo studies have corroborated CGRP as a mediator of Th2-type immune responses [43]. The significance of CGRP’s role in shifting toward a Th2 immune response is not fully understood.
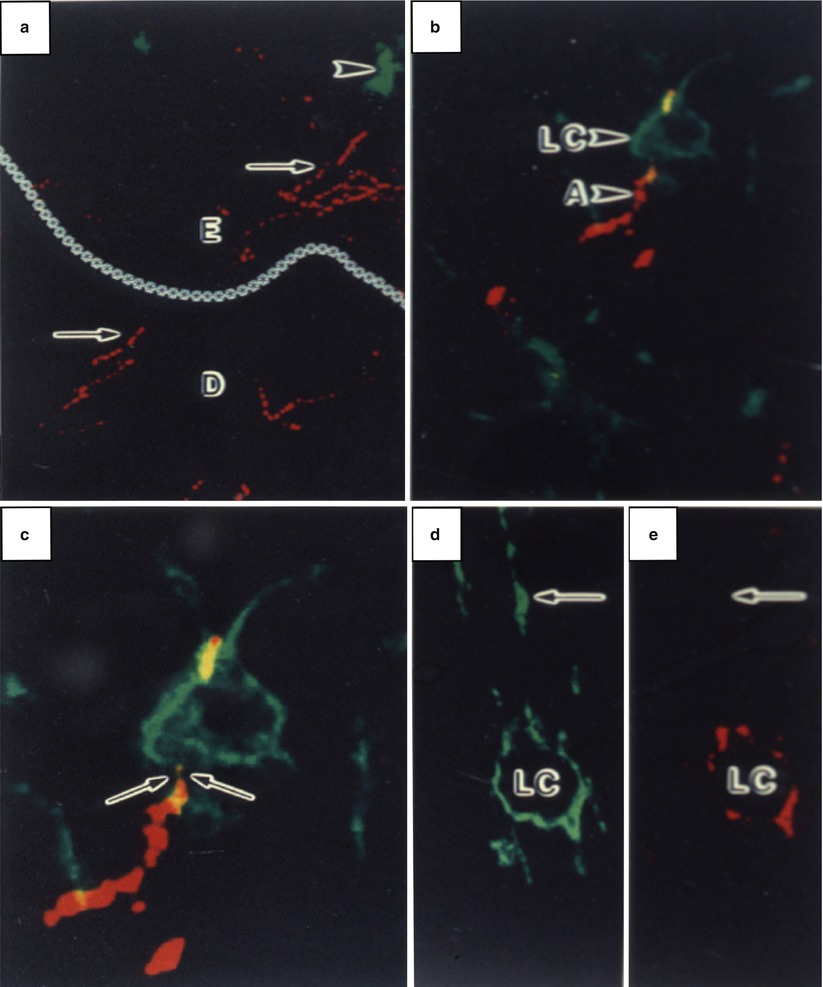
Fig. 12.1
Laser confocal scanning microscopy illustrating the anatomic association between Langerhans cells (LCs) and CGRP-containing nerve axons (A; arrows) in the dermis (D) and epidermis (E) (dermal-epidermal junction demarcated by asterisks). LCs, shown in green, were stained with an antibody against CD1a. Nerves, shown in red, were stained with an antibody against CGRP. (a) CGRP-positive axons in the epidermis and dermis (arrows) and CD1a-positive LCs (arrowheads). (b, c) Apparent contact of CGRP-positive axon and a LC. (d, e) Single channel analysese of LC CD1a reactivity (D) and CGRP staining (E). (Reprinted with permission from Hosoi et al. [40])
CGRP is a mediator of neurogenic vasodilatation and a modulator of keratinocyte proliferation and cytokine production [2]. Human primary dermal microvascular endothelial cells (pDMECs) express mRNA for CGRP and adrenomedullin receptors, and recent investigations found that CGRP inhibits lipopolysaccharide-induced production of various chemokines, including IL-8, MCP-1 and GRO-α by pDMECs, as well as by human microvascular endothelial 1 cells (HMEC-1) [44]. These cells were found to express all components of the CGRP receptors at the mRNA level and the effects on chemokine release could be blocked by the type I CGRP receptor antagonist CGRP8–37 [44]. CGRP inhibits NF-kB activation in the HMEC-1 cell line [44], and inhibition of NF-kB signaling was found to suppress the production of these chemokines [44]. Accordingly, CGRP treatment also suppresses neutrophil and monocyte chemoattraction by LPS-stimulated HMEC-1 cells [44]. These experiments suggest a role for CGRP in endogenous anti-inflammatory pathways, in addition to its effects on the adaptive immune response [31]. There is also some in vitro and in vivo experimental evidence that CGRP inhibits inflammation via a pathway that involves rapid upregulation of the inducible cAMP early repressor (ICER) [45]. ICER is a repressor protein that, in dendritic cells, suppresses LPS-induced transcription of genes encoding the inflammatory molecules TNF-α and CCL4, both of which share the quality of having a CRE-kB promoter element [45]. CGRP did not attenuate the expression of genes such as the one that encodes CXCL1 that lack this element [45]. These findings were found to occur in the absence of IL-10, a well-known suppressor of inflammation, suggesting that CGRP suppresses immune responses by cAMP-induced activation of intracellular kinases such as protein kinase A (PKA), providing an alternative to the NF-kB and MAPK pathways as a route by which interference with the downstream effects of toll-like receptor (TLR) signaling ultimately attenuates inflammation [45].
CGRP is involved in ultraviolet radiation (UVR)-induced immunosuppression. In the low dose model of UVB-induced immunosuppression (sensitization to a hapten is impaired at the irradiated site) UVR releases CGRP from sensory neurons to locally impair contact hypersensitivity responses [46]. The release of CGRP after UVR may be triggering mast cells to release stored TNF-α, which in turn down-regulates Langerhans cell density and function to impair CH induction [47]. CGRP also contributes to the high-dose or systemic model of UVR-induced immunosuppression (sensitization at a non-irradiated site is impaired) as suppression of CHS responses are inhibited when mice are pretreated with CGRP antagonists [48].
Proopiomelanocortin Peptides
The skin is a source of neuroendocrine hormones of the proopiomelanocortin (POMC) family, termed melanocortins (MCs). POMC hormones include α, β, and γ-melanocyte stimulating hormone (MSH). These are derived from POMC along with adrenocorticotrophic hormone (ACTH) and endorphins. POMC is predominantly synthesized in the pituitary gland; however, it is also present in the skin, expressed by melanocytes, keratinocytes, Langerhans cells, endothelial cells, mast cells and fibroblasts [10]. The cutaneous melanocortin system is well characterized [49] with melanocortin receptor (MC-R) expression in nearly all cell types, including fibroblasts, adipocytes, endothelial cells and mast cells [50]. MC-1R is the most prevalent amongst the five melanocortin receptors, with high affinity for α-MSH and ACTH.
POMC-derived peptides influence several processes including melanogenesis and skin immunity, and are also suggested to have a role in the hair cycle, sebum and eccrine gland function and epidermal proliferation [50]. The known functions of melancortins in cutaneous biology continue to expand with potential clinical applications. Human sebocytes increase lipid droplet formation in response to MCs, suggesting acne vulgaris may be affected. Evidence suggests MCs are involved in keratinocyte proliferation and differentiation, with potential repercussions in regenerative processes [49]. In addition, although the mechanism is not completely understood, it is believed that MSH induces melanocyte proliferation and melanogenesis through engagement of the MC1-R, serving as a substrate or promoter of the enzyme tyrosinase [19].
POMC mRNA and related peptides such as α-MSH and ACTH are upregulated by UVR. Mid-range UVR [UVB radiation (280–320 nm)] upregulates ACTH, α-MSH and MC-1R in epidermal melanocytes and increases MC-1R expression in cultured normal and malignant melanocytes and keratinocytes, the latter associated with increased melanogenesis in cell culture and in vivo [9, 49]. The melanogenic and dendritogenic effects of α-MSH and ACTH on the skin and follicular melanocytes may correlate with tanning and hair color, respectively [49].
POMC-derived peptides play a role in immunity and inflammation, especially α-MSH. α-MSH is anti-inflammatory and may be involved in host defense. More specifically, it has been shown to inhibit neutrophil migration and block the production of inflammatory cytokines, including IFN-γ, TNF-α, IL-1 and IL-6, by macrophages [51]. In studies of human microvascular endothelial cells, α-MSH has been found to suppress lymphocyte adhesion and inhibit lipopolysaccharide-mediated upregulation of ICAM-1, CAM and E-selectin receptors [51]. α-MSH also inhibits contact hypersensitivity and induces hapten-specific tolerance through IL-10 upregulation in a murine model [52]. Recent experiments demonstrated that α-MSH encourages dendritic cells to induce a T regulatory cell immune response, which in turn suppressed the proliferation of and cytokine secretion from Th17 cells obtained from individuals with psoriasis [53].
α-MSH has also been found in a murine model to augment antitumoral immunity by enhancing CD8+ T cell cytolytic activity via interactions at the MC-1R [54]. These studies demonstrated that α-MSH-stimulated CD8+ T cells were associated with significant in vitro as well as in vivo anti-tumor effects [54]. In addition, the anti-TNF and antimicrobial effects of α-MSH suggest it may reduce replication of the human immunodeficiency virus (HIV), because TNF promotes HIV replication [55]. Furthermore, its anti-inflammatory properties in animal models of hepatic inflammation and arthritic processes suggest that it may be an important anti-inflammatory agent in the treatment of inflammatory diseases in humans [56–58].
Catecholamines
Sympathetic fibers of the autonomic nervous system travel with sensory nerve fibers and as single fibers to innervate blood vessels, hair follicles and sweat glands [5]. Keratinocytes also synthesize catecholamines [9, 59]. Catecholamines inhibit antigen presentation in Langerhans cells through the β2 adrenergic receptor [60]. The sympathetic neurotransmitter norepinephrine and the co-transmitter adenosine triphosphate (ATP) have been shown to act synergistically to induce IL-6 production by human dermal microvascular endothelial cells [61]. As IL-6 is important in the differentiation of Th17 cells, this finding suggests a possible link between stress-associated activation of the sympathetic nervous system and Th17-cell-mediated immune responses [61]. This might help to explain the role of stress in exacerbating psoriasis and other skin disorders in which Th17 mechanisms play a role [61, 62].
Interestingly, evidence exists that norepinephrine plays a role in vivo in enhancing CHS through involvement in the trafficking of skin dendritic cells to draining lymph nodes, possibly through effects on α-adrenergic receptors [63]. Thus, the effects of catecholamines on skin immunity may be complex, with the outcome dependent upon factors such as the timing of exposure and the presence of other regulatory factors.
Adenosine 5´-Triphosphate (ATP)
The nucleotide ATP is a ubiquitous carrier of energy involved in countless cellular processes. Several lines of evidence suggest extracellular ATP participates in inflammatory and regenerative responses in the skin and affects melanocyte and Langerhans cell function [64]. ATP plays a role in the nociceptive signaling pathways following cutaneous cell injury [65] and may contribute to the pathophysiology of inflammatory skin conditions such as rosacea by augmenting the production of inflammatory mediators by endothelial cells [66]. The human microvascular endothelial cell line HMEC-1 expresses a variety of ligand-gated purinergic P2X receptors and G protein-coupled purinergic P2Y receptors which, when activated with ATP or the long-lived ATP analog ATPγS, was associated with increased production of the inflammatory cytokine IL-6 and the chemokines IL-8, MCP-1 and GRO-α [31, 66] Furthermore, in vitro studies revealed that tetracycline inhibited ATPγS- and TNFα-induced release of GRO-α and IL-8 by HMEC-1 cells and human dermal microvascular endothelial cells, a finding that may account, in part, for the beneficial effects of these agents in treating various inflammatory disorders [67].
Purinergic agonists also appear to enhance Langerhans cell antigen presenting function when exposure occurs along with activation by an additional signal (lipopolysaccharide) [68]. In this regard an ATP analog and P2X7R agonist 2′(3′)-O-(4-benzoylbenzoyl) ATP (BzATP) was shown to induce Th17-type immune responses in lesional and nonlesional skin from patients with psoriasis [69]. The authors interpreted this as due to action at the P2X7 receptor, which is coupled to GS-dependent intracellular changes in cAMP and its downstream effectors; however, BzATP also has high potency at P2X1R [70] as well as activity at other P2 receptors [15]. Investigators observed that this response was carried out via antigen-presenting-cell-dependent mechanisms and was mediated by the pro-inflammatory cytokines IL-6, TNF-α and IL-1β and IL-23 [69]. Among other pro-inflammatory activities, these cytokines promote Th17 cell differentiation, supporting the apparent role of Th17 cells in the pathogenesis of psoriasis [69]. Furthermore, IL-6 secretion from endothelial cells is upregulated synergistically in response to ATP and norepinephrine signaling [61], and may contribute to the pathogenesis of psoriasis by inhibiting regulatory T cell functions [69]. These data may better explain the relationship between sympathetic nervous system activity and psoriasis flares. The function of ATP in the neuropeptide milieu of epidermal and dermal cell types and sensory nerves in the skin remains the subject of ongoing investigations.
Neuropeptide Y (NPY)
NPY is a 36-amino-acid peptide identified predominantly in periarteriolar nerve fibers in the dermal plexuses and, epidermal basal cells as well as sweat glands, sebaceous glands and hair follicles. It also appears to be produced by Langerhans cells [71]. NPY causes vasoconstriction and may also play a role in eccrine sweat production. It also reportedly has antimicrobial properties [72].
Catestatin
Catestatin is a 21-amino-acid neuropeptide derived from chromogranin A, which is a neuroendocrine pro-hormone and a member of the granin family of peptides [31]. This family of proteins is found within secretory vesicles of nerve, endocrine and immune cells that release antimicrobial peptides derived from chromogranin A [73]. Catestatin is upregulated in response to injury [74]. It displays antimicrobial properties against cutaneous pathogens including gram-positive and negative bacteria, yeast, and fungi [74, 75], and also acts as a chemoattractant for monocytes [76]. Furthermore, catestatin was observed to stimulate keratinocytes to migrate and produce IL-8, suggesting a role in immunomodulation [77]. Additionally, catestatin has been shown to induce human mast cell migration, degranulation, and pro-inflammatory cytokine and chemokine production [75]. This may contribute to the known role that mast cells play in cutaneous neurogenic inflammation.
Neurogenic Inflammation
Cutaneous neurogenic inflammation implies the role of nerves in cutaneous inflammation. The neuropeptides secreted by sensory nerves evoke an inflammatory response referred to as neurogenic inflammation. The axon-reflex model implies that tissue injury triggers a signal to the dorsal root ganglion toward the central nervous system (orthodromic response) with return of the signal from branch points in the reverse direction (antidromic signaling) to exert effects at the local level. The orthodromic response transmits pain whereas the antidromic response leads to the release of neuropeptides in the innervated tissue [12].
Various inflammatory cells are known to express neuropeptide receptors. Neutrophils express receptors for SP including NK1, NK2 and NK3 [78]. SP reportedly upregulates neutrophil expression of COX-2 and PGE2 [78]. Furthermore, mast cells express receptors for various neuropeptides including VIP, NYP, SP, CGRP, POMC, galanin, neuromedin U, PACAP, neurotensin and CRH [78]. Although located throughout the body, mast cells localize near blood vessels, nerve fibers and lymphatic vessels [31]. More specifically, mast cells and nerves engage in a bidirectional interaction in which degranulation provokes membrane depolarization of sympathetic neurons [78]. Pre-treatment of mast cells with selective antagonists of substance P and CGRP in a murine model was observed to decrease neuropeptide-associated degranulation, supporting the role of this interaction in neurogenic inflammation [78].
The most prominent neuropeptides in UVR-induced neuroinflammation are SP and CGRP. SP and CGRP mediate vasodilatation and SP and NKA are responsible for plasma extravasation [22]. SP is well known to provoke erythema and edema by mast cell-dependent and independent pathways [10]. The flare is the result of the antidromic arm of the axon reflex leading to the local release of neuropeptides such as SP and CGRP [20]. Both H1 receptor antagonism and local anesthetic injection have been shown to reduce the spread of the flare response to SP in human skin, suggesting a prominent role of histamine in neurogenic inflammation [79].
Cutaneous/Dermatologic Diseases
The relationship between neuropeptides and dermatologic diseases has been explored for decades. In inflammatory processes, the neuro-immuno-endocrine-cutaneous nexus participates in the trigger and maintenance of inflammation in healthy and pathologic skin. Understanding the role of neurotransmitters and their receptors may lead to the identification of novel therapeutic targets for the treatment of several common cutaneous diseases.
There are several lines of evidence suggesting a neurogenic component in dermatologic disease. Neuropeptides may induce or alleviate urticarial symptoms, hypersensitivity reactions and rosacea, and may play a role in the pathophysiology of pruritus, psoriasis, atopic dermatitis, alopecia areata, vitiligo and nodular prurigo [2, 12]. Several of these conditions are discussed below.
Urticaria
Urticaria presents as transient swellings or wheals caused by plasma leakage. The primary effector cell in the pathogenesis of urticaria is the mast cell [5]. Neuropeptides such as SP, VIP and somatostatin active mast cells to secrete histamine and other mediators that induce urticaria and mediate the late-phase response [5, 80]. SP and CGRP exert effects in cases of chronic urticaria and ACTH is present in the cutaneous mast cells of patients with urticaria pigmentosa [81, 82]. The expression of the POMC gene in mast cells suggests that α-MSH may be contributing to the cutaneous hyperpigmentation seen in patients with urticaria pigmentosa [83]. These studies suggest an interrelationship between the cutaneous nervous system and mast cells in the pathophysiology of urticaria.
Pruritus
Pruritus is one of the most common symptoms encountered in dermatology and significantly impacts quality of life. Itch may be peripheral (dermal or neuropathic) or central (neuropathic, neurogenic or psychogenic) in origin. Neuropathic itch originates at any point along the afferent pathway and is the result of damage to the nervous system whereas neurogenic itch is induced centrally. In the skin, itch is induced by the stimulation of specialized C-fibers by various pruritogens. This is followed by the release of neuropeptides from the cleavage of type-2 proteinase-activated receptors (PAR-2) by tryptase to release histamine, CGRP and SP [84].
It is important to understand the underlying mechanisms to provide effective management and treatment. Multiple cutaneous mediators such as histamine, prostanoids, cytokines and kinins induce pruritus [80]. Pruritus-inducing mechanisms of these mediators include nerve fiber sensitization and receptor stimulation, direct pruritogenic effects and mast cell activation [7]. Histamine is well known for its pruritic effects, especially in urticaria. However, in conditions such as atopic dermatitis the inability of antihistamines to eliminate itch suggest that other mediators are involved as well. Intradermal injection of SP, VIP and somatostatin evokes pruritus; however, CGRP does not have pruritogenic effects in humans [80]. SP is released from C-fiber nerve terminals by the action of mast cell tryptase on PAR-2 in the terminals to directly cause itching and by inducing mast cells to release histamine. Lesional skin in chronic pruritus demonstrates a predominance of dermal SP-containing nerve fibers relative to sympathetic nerve fibers [85]. Additional studies have shown that the pruritic lesional skin of patients with psoriasis contained an increased number of both SP-positive nerves and NK2 receptor-positive cells. These data suggest that antagonizing the SP receptors NK1 and NK2 may be useful in treating patients with pruritic skin disorders [86].
Experimental evidence suggests that the itch circuitry responsible for chronic pruritus may be under tonic inhibitory control by specific types of sensory neurons that are sensitive to mechanical stimulation [87]. These effects are thought to be mediated by members of the TRPV1 family of ion channels [87]. In a murine model, toxin-mediated ablation of GRP- and neuromedin B receptors was associated with abolition of scratching behavior after intradermal application of various pruritogens [88, 89].
There are continuing new developments surfacing in the understanding of the pathogenesis of pruritus including the identification of IL-31 as a role-player in inflammation and pruritus [90]. IL-31 is a known product of mast cells as well as Th2 cells [88]. Interestingly, blood levels of IL-31, CGRP, and SP were recently found to be significantly higher in psoriasis patients than in a control group while the level of CRH was reduced [82]. After 20 treatments with narrow-band UVB radiation, the IL-31 level was significantly reduced, suggesting a possible role in some manifestations of psoriasis [91]. IL-31 levels are also reportedly increased in the serum of patients with chronic urticaria and atopic dermatitis [88]. IL-31 receptors have been found on DRG neurons, suggesting that IL-31 may directly activate these sensory nerve fibers, and perhaps offering an explanation for its role in inducing the sensation of itch [88].
Atopic Dermatitis
Atopic dermatitis (AD) is characterized by cutaneous hyper-reactivity to nonspecific stimuli leading to a cycle of pruritus, scratching and further worsening of skin lesions [92]. AD is characterized by increased amounts of Th2-derived cytokines [93], as well as an increased density of nerve fibers and neuropeptide levels in the various stages of skin lesions seen in this condition [94]. The diameter of these fibers are larger than those in nonatopic control subjects possibly due to keratinocyte-derived, nerve growth factor (NGF) [95]. The free nerve endings in the skin of atopics also lack a surrounding sheath of Schwann cells suggesting an active state of excitation [94].
SP is a potent pruritogenic neuropeptide in AD that, along with CGRP, is down regulated by phototherapy [96, 97]. An increase in nerve fibers containing SP and CGRP is often observed although Fantini et al. demonstrated a decrease in SP and an increase in VIP levels in chronic lesions of AD [98, 99]. In lesional skin of AD and nummular dermatitis patients, SP and CGRP, but not VIP, fibers in the dermis are elevated compared to nonlesional controls, and are likely maintained by an increased number of mast cells [100]. Intradermal SP increases nitric oxide and enhances SP-induced pruritus while acute stress triggers SP-induced cutaneous mast cell degranulation [7]. Elevated VIP was found to be present in eczema [101].
Plasma levels of NGF and VIP are reportedly higher in patients with AD than in psoriatic patients and healthy controls [102]. SP plasma concentrations were higher in a specific sub-group of AD patients with “extrinsic AD,” a definition that referred to patients who presented with IgE-mediated sensitization to common food and/or aeroallergens based on skin prick tests and/or elevated total IgE serum levels [102]. Investigators using a skin model based on the coculture of human dermal fibroblasts, keratinocytes and porcine DRG neurons found that neural-derived CGRP enhanced both keratinocyte proliferation and epidermal thickness, phenotypic changes consistent with the atopic phenotype. This isolate skin cell model reportedly allowed investigators to study the interaction of nerve and skin cells under defined conditions in which the effects of porcine neurons, which are most similar to their human counterparts, could be assessed. Interestingly, a thicker epidermis was seen when keratinocytes came from atopic skin [103]. These results were interpreted as demonstrating that epidermal nerve endings enhance the morphologic changes associated with the atopic phenotype [103]. Pharmacologic inhibition of CGRP signaling, but not SP signaling, reversed the atopic phenotypic changes. The strong effects of neuronal integration appeared to be mediated by upregulation of CGRP receptor mRNA components in both healthy and atopic models. These data may suggest a relationship between the skin barrier dysfunction characteristic of AD and abnormal CGRP signaling [103].
Allergen-induced late-phase reactions in patients with AD sensitive to a known allergen have been associated with infiltrates of inflammatory cells expressing CGRP and VEGF, suggesting a role of these compounds in mediating the erythema and inflammation that may be associated with allergic inflammation [104]. In situ hybridization revealed that neutrophils and T cells were the predominant CGRP-producing cells in late-phase edema [104]. Studies comparing these results with patients that have allergic diatheses, but not AD, do not appear to have been performed.
Evidence suggests that there may be a relationship between atopic dermatitis and its associated psychological symptoms, including anxiety and depression, which may be accounted for by cutaneous neuropeptide activity. One study demonstrated that SP and NGF are useful serum markers of disease activity in patients with AD, which was evaluated clinically using three different scoring systems [105]. Indeed, in another study NGF-reactive cells were detected more abundantly in epidermal and dermal AD-involved skin [106]. NPY-positive cells were also detected in greater abundance in the epidermis of patients with AD compared to healthy controls. Investigators reported that pruritus in patients with AD was positively correlated with state and trait levels of anxiety, and hypothesized that anxiety may induce pruritus through NPY- and NGF-dependent mechanisms in the skin that are believed to involve mast cells, fibroblasts, lymphocytes and eosinophils [106].
Recently, inherited mutations in filaggrin have been appreciated for the role they may play in AD pathogenesis. Filaggrin (filament-aggregating protein) and its precursor profilaggrin are among the main components of the F-type keratohyaline granules that comprise the stratum granulosum, and as such, filaggrin plays an integral role in maintaining skin hydration [107]. The relationship of filaggrin to AD appears to involve the role it plays in maintaining epidermal barrier function, the integrity of which is known to be lost in many patients with AD. There are more than 45 known mutations in the filaggrin-encoding gene, FLG, at least four of which are believed to contribute to AD pathogenesis [107, 108]. The most common loss-of-function mutation in FLG is found in almost one-third of European Caucasian patients [109]. However, the four recurrent FLG mutations associated with AD only have an estimated combined allele frequency of approximately 7–10 % [108], and at least 50 % of AD patients have no FLG defects [107]. Furthermore, patients with FLG mutations may develop other conditions, such as ichthyosis vulgaris, without developing AD [107]. Recent evidence suggests that the relationship of preexisting FLG mutations and AD development may be related to the expression of immune modulators such as thymic stromal lymphopoietin (TSLP), which is known to be involved in initiating allergic inflammatory responses [110]. Interestingly, keratinocytes that differentiate in the presence of Th2 cytokines show a lower filaggrin protein expression, independent of underlying FLG mutations [109]. The established role of neuropeptides in regulating the aberrant cutaneous immune responses seen in AD suggests that the pathogenesis of this disorder is multi-faceted and involves a complex interaction between nerve-derived signaling molecules, Th2-type immune responses and abnormal skin barrier function.
Psoriasis
Psoriasis is a multifactorial disease characterized by symmetric plaque lesions. The possibility of a neurogenic component is supported by the temporal onset or exacerbation of lesions with emotional stress, the appearance of lesions at sites of injury or trauma (Koebner phenomenon – believed to be initiated by the release of proinflammatory neuropeptides in traumatized skin) and by observations that lesions resolve in areas of denervation or following central nervous system lesions such as ischemic stroke [80, 111].
NPs and sensory nerve density are increased in lesional skin [80]; however, levels of SP and VIP vary in psoriasis. Elevated levels of both SP and VIP [112], normal SP and elevated VIP [101] and increased SP and normal VIP levels [113] have all been demonstrated in psoriatic lesions. There are conflicting reports of CGRP expression in lesional and nonlesional skin compared to normal controls. Moreover, VIP expression is reported to be increased, yet a statistical difference is not consistently observed between lesional and normal skin [111]. Serum CGRP and VIP are both reportedly elevated in psoriatic individuals. Treatment with anthralin or psoralen plus long-wave UVR [UVA radiation (320–400 nm)] (PUVA) led to a decrease in serum VIP while CGRP remained elevated [114]. Below we discuss evidence that VIP may be involved in biasing immune responses toward the Th17 pole, which suggests the potential relevance of these findings to enhancing the current understanding of psoriasis pathogenesis. Compared to psoriatic lesions, a lower level of SP is seen in lichen planus and lichen simplex chronicus while higher levels are observed in spongiotic dermatoses [113]. Serum beta-endorphin is elevated in psoriatic patients and is likely produced by inflammatory cells within psoriatic plaques [115]. PACAP-38 is also increased in lesional psoriatic skin and recent evidence suggests NGF is involved in the pathogenesis of psoriasis [28, 80]. Keratinocyte hyperproliferation is a histologic feature in psoriasis and NGF is suggested to induce keratinocyte proliferation and prevent apoptosis [111]. SP reportedly upregulates keratinocyte-derived IL-32 and mast-cell-derived tryptase in psoriatic lesional skin [116]. Additional evidence for neuropeptide-mediated mast cell involvement in psoriasis pathogenesis comes from investigations revealing that SP stimulates mast cells to increase CRH receptor (CRHR-1) expression, which in turn enhances NK1 gene transcription, mediating SP signaling [117]. Overstimulation with CRH, as in chronic stress, reportedly leads to down-regulation of CRHR-1 on the surface of mast cells, which may account for previous observations of decreased CRHR-1 gene expression in psoriatic lesional skin [117]. Together, these data may account for the relationship of stress-induced activation of the hypothalamic-pituitary-adrenal axis to psoriasis flares.
Novel murine models of psoriasis have enabled further studies of the role of neurocutaneous immunity in the pathogenesis of this disorder. The KC-Tie2 murine model is engineered to overexpress the angiopoietin receptor Tie2 in keratinocytes [32]. Tie2 receptor activation is associated with reactive oxygen species generation, which may mediate keratinocyte proliferation via VEGF production and angiogenesis [118]. KC-Tie2 mice spontaneously develop features characteristic of human psoriasis including increased numbers of dermal CD4+ T cells, epidermal CD8+ T cell infiltrates and pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-1α, IL-6, IL-12, IL-22, IL-23 and IL-17 [118]. In this model, systemic administration of the T-cell calcineurin inhibitor cyclosporin A was associated with significant improvement in the psoriasiform phenotype and a reduction in lesional T cells and their associated pro-inflammatory cytokines, an observation consistent with the role that T lymphocytes are believed to play in psoriasis pathogenesis [118]. In these experiments, antigen presenting cells remained high in lesional skin, which may have accounted for incomplete resolution of the disease phenotype [118]. In fact, clodronate-liposome-mediated depletion of antigen presenting cells in KC-Tie2 mouse skin was associated with resolution of the acanthotic skin phenotype, decreased dermal angiogenesis and normalization of the levels of multiple pro-inflammatory cytokines.
Additional investigations using the KC-Tie2 model revealed that denervation of cutaneous sensory nerves led to significant drops in lesional dendritic and CD4+ T cells, reminiscent of denervation in human patients with diseases of the nervous system. However, administration of an SP agonist prevented decreases in CD11c+ and CD4+ cells, but had no effect on acanthosis [119]. Interestingly, administration of CGRP reversed the improvement in acanthosis and prevented the denervation-mediated decrease in numbers of CD4+ cells [119]. Perhaps the relationship of sensory nerve activation to disease activity, i.e. secondary to external stimulation such as pressure, may partially contribute to the aforementioned Koebner phenomenon [119].
The imiquimod mouse model of psoriasis uses a toll-like receptor (TLR) 7 agonist to induce a psoriatic phenotype [120]. Investigators using this model to study the pathogenetic mechanisms of psoriasis found that IL-23-mediated activation of IL-17 and IL-22 cells was requisite in triggering the psoriasiform phenotypic changes characteristic of this model [120]. Additional investigations found that IL-22, a cytokine produced by Th1, Th17 and Th22 cell populations as well as by innate immune cells, is required for the imiquimod-induced psoriasiform inflammatory changes characteristic of this murine model, supporting the known role of these novel T cell populations in triggering psoriasis [121]. Recent evidence suggests that nociceptive sensory neurons expressing specific ion channels including TRPV1 are essential for driving this inflammatory through stimulating dermal dendritic cell to produce IL-23 which, in turn, induces dermal gamma-delta T cells to produces IL-23-dependent inflammatory cytokines [122]. Accordingly nociceptive sensory neuron loss was associated with a reduction in both local swelling and inflammatory infiltrates [122]. Furthermore, imiquimod treatment was associated with increased levels of IL-17A, IL-17F and IL-22, consistent with previous reports demonstrating the role of these cytokines in psoriasiform inflammation [122]. Finally, recent experiments have demonstrated that cutaneous denervation results in the loss of imiquimod’s ability to induce the psoriatic phenotype, highlighting the essential role of cutaneous nerves and the neuropeptides they secrete in triggering this skin disorder [123].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


