Introduction938
INTRODUCTION
NEUTROPHIL INFILTRATES
Neutrophils
Diseases with neutrophilic infiltrates
Epidermal neutrophilic infiltrates
Impetigo
Toxic shock syndrome
Dermatophytoses
Chromomycosis
Sporotrichosis
Milker’s nodule
Orf
Yaws
Subcorneal pustular dermatosis
Acute generalized exanthematous pustulosis
Pustular psoriasis
Psoriasis
Reiter’s syndrome
Palmoplantar pustulosis
Pustular eruption of ulcerative colitis
Infantile acropustulosis
Erythema neonatorum toxicum
Transient neonatal pustular melanosis
Pemphigus foliaceus
IgA pemphigus
Miliaria pustulosa
Acute generalized pustulosis
Glucagonoma syndrome
Halogenodermas
Verruciform xanthoma
Adult T-cell leukemia/lymphoma
Dermal neutrophilic infiltrates
Infections and infestations
Ecthyma
Erysipelas
Erysipeloid
Cellulitis
Blastomycosis-like pyoderma
Erosive pustular dermatosis of the scalp
Mycobacterium ulcerans infection
Other atypical mycobacterial infections
Erythema nodosum leprosum
Chancroid
Granuloma inguinale
Kerion
Actinomycosis
Nocardiosis
Mycetoma
Acute cutaneous leishmaniasis
Secondary syphilis
Bite reactions (fleas, ticks, and fire-ants)
Neutrophilic dermatoses
Periodic fever syndromes
Sweet’s syndrome
Pustular vasculitis of the hands
Bowel-associated dermatosis–arthritis syndrome
Rheumatoid neutrophilic dermatosis
Acute generalized pustulosis
Behçet’s syndrome
Pyoderma gangrenosum
Acute vasculitides
Hypersensitivity vasculitis and variants
Septic vasculitis
Erythema elevatum diutinum
Granuloma faciale
Polyarteritis nodosa
Subepidermal blistering diseases
Dermatitis herpetiformis
Bullous systemic lupus erythematosus
Cicatricial pemphigoid
Ocular cicatricial pemphigoid
Localized pemphigoid
Linear IgA bullous dermatosis
Epidermolysis bullosa acquisita
Deep lamina lucida pemphigoid
Folliculitides
Bacterial and fungal folliculitis
Secondary syphilis
Perforating folliculitis
Miscellaneous conditions
Neutrophilic urticaria
Polymorphic light eruption
Cutis laxa (early stage)
Eruptive xanthoma
Reticulohistiocytoma
Anaplastic large cell lymphoma
Erythropoietic protoporphyria
Neutrophilic eccrine hidradenitis
Familial Mediterranean fever
Congenital erosive and vesicular dermatosis
Neutrophilic figurate erythema
Still’s disease
Subcutaneous neutrophil infiltrates
Infective panniculitis (causes as for dermal infiltrates – see above); erythema nodosum leprosum
Factitial panniculitis
Pustular panniculitis of rheumatoid arthritis
α1-Antitrypsin deficiency
EOSINOPHIL INFILTRATES
Eosinophils
Diseases with conspicuous eosinophils
Vesiculobullous diseases
Dermatitis herpetiformis (late lesions)
Bullous pemphigoid
Bullous arthropod bite reaction
Pemphigus vegetans
Pemphigoid gestationis
Eosinophilic spongiosis
Erythema neonatorum toxicum
Disorders of blood vessels
Urticaria
Hypersensitivity vasculitis (some cases)
Eosinophilic vasculitis
Allergic granulomatosis (Churg–Strauss)
Juvenile temporal arteritis
Angiolymphoid hyperplasia with eosinophilia
Kimura’s disease
Infections/infestations
Parasitic infestations
Dermatophytes (uncommon)
Miscellaneous conditions
Hypereosinophilic syndrome
Wells’ syndrome
Eosinophilic annular erythema
Dermatitis cruris
Pachydermatous eosinophilic dermatitis
Incontinentia pigmenti
Allergic contact dermatitis
Drug reactions
Dermal hypersensitivity
Eosinophilic pustular folliculitis
Eosinophilic pustulosis
Eosinophilic panniculitis
Eosinophilic fasciitis
Eosinophilic, polymorphic and pruritic eruption of radiotherapy
Tumors
Langerhans cell histiocytosis
Tumor-like eosinophilic granuloma
Juvenile xanthogranuloma
Eosinophilic variant, lymphomatoid papulosis
Squamous cell carcinoma
Keratoacanthoma
Malignant melanoma (rare)
WELLS’ SYNDROME (EOSINOPHILIC CELLULITIS)
Histopathology23.67. and 68.
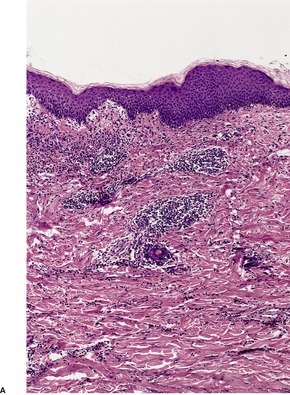
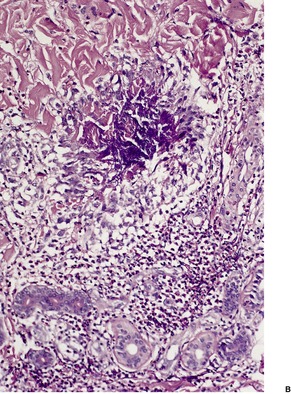
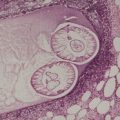
Fig. 40.1
Electron microscopy
HYPEREOSINOPHILIC SYNDROME
Histopathology111
PACHYDERMATOUS EOSINOPHILIC DERMATITIS
Histopathology
DERMAL HYPERSENSITIVITY REACTION
Histopathology
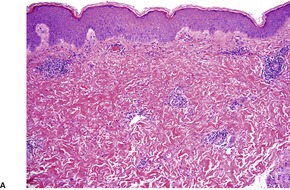
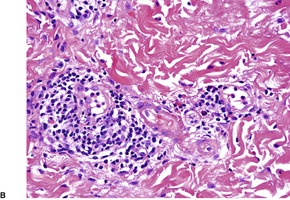
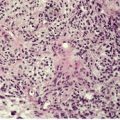
Fig. 40.2
EOSINOPHILIC PUSTULOSIS
Histopathology
PLASMA CELL INFILTRATES
Plasma cells
Plasma cell hyperplasias
PLASMACYTOMAS AND MULTIPLE MYELOMA
Multiple myeloma
Histopathology
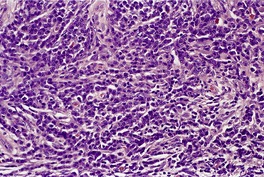
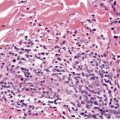
Fig. 40.3
Electron microscopy
Cutaneous disorders associated with paraproteinemias
WALDENSTRÖM’S MACROGLOBULINEMIA
Histopathology193
Electron microscopy
CUTANEOUS AND SYSTEMIC PLASMACYTOSIS
Histopathology
PLASMACYTOSIS MUCOSAE (INCLUDING ZOON’S BALANITIS/VULVITIS)
Histopathology229
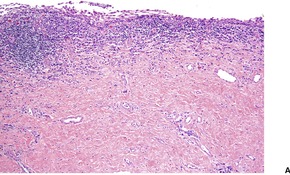
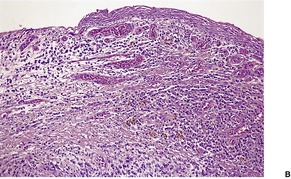
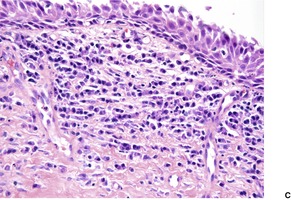
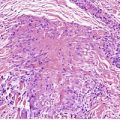
Fig. 40.4
Electron microscopy227
CASTLEMAN’S DISEASE
Histopathology
ANGIOPLASMOCELLULAR HYPERPLASIA
MAST CELL INFILTRATES
Mast cells
Mast cell hyperplasias
MASTOCYTOSIS
*From Valent et al (Leuk Res 2001; 25: 603–625).
Cutaneous mastocytosis (CM)
Maculopapular CM
Diffuse CM
Mastocytoma of skin
Indolent systemic mastocytosis (SM)
Smoldering SM
Isolated bone marrow mastocytosis
Systemic mastocytosis with an associated clonal hematological non-mast cell lineage disease (MCD-AHNMD)
Aggressive systemic mastocytosis
With eosinophilia
Mast cell leukemia (MCL)
Aleukemic MCL
Mast cell sarcoma
Extracutaneous mastocytoma
Urticaria pigmentosa
Solitary mastocytoma
Diffuse cutaneous mastocytosis
TMEP
Systemic mastocytosis
Malignant mast cell disease
Histopathology270
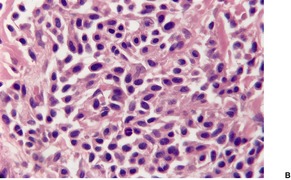
Fig. 40.5
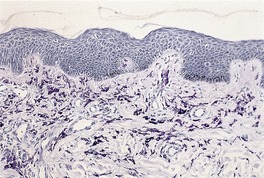
Fig. 40.6
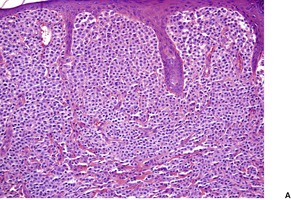
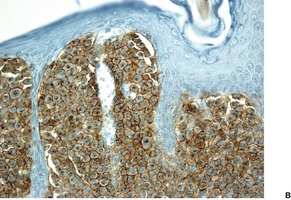
Fig. 40.7
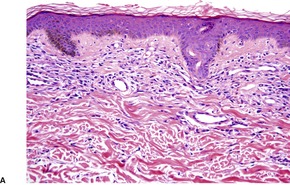
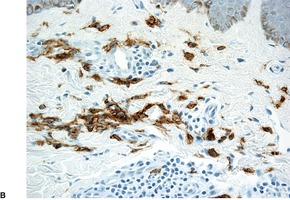
Fig. 40.8
Electron microscopy
BRACHIORADIAL PRURITUS
Histopathology
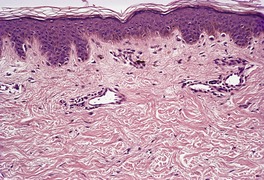
Fig. 40.9
HISTIOCYTIC INFILTRATES (NON-LANGERHANS CELL)
Histiocytes
Cell type
Features
Macrophage
Dermal dendrocyte
Indeterminate cell
Langerhans cell
*This classification is based on the Histiocyte Society/WHO classification of 1997, and excludes non-cutaneous diseases. (Med Pediatr Oncol 1997; 29: 157–166).
Disorders of varied biological behavior
Dendritic cell-related
Langerhans cell histiocytosis
Juvenile xanthogranuloma and related disorders
Solitary histiocytomas with dendritic cell phenotypes
Macrophage-related
Hemophagocytic syndromes (primary and secondary)
Rosai–Dorfman disease
Solitary histiocytoma with macrophage phenotype
Malignant disorders
Monocyte-related (various leukemias)
Dendritic cell-related histiocytic sarcoma
Macrophage-related histiocytic sarcoma
Cell type
Histiocytoses
Vacuolated
Xanthomatized
Spindle-shaped
Scalloped
Xanthoma disseminatum
Oncocytic
Multicentric reticulohistiocytosis
Mixed
Predominantly affecting skin
Juvenile xanthogranuloma, cephalic histiocytosis and others
Skin + major systemic involvement
Xanthoma disseminatum
Primarily involve extracutaneous sites
JUVENILE XANTHOGRANULOMA
Histopathology483. and 541.
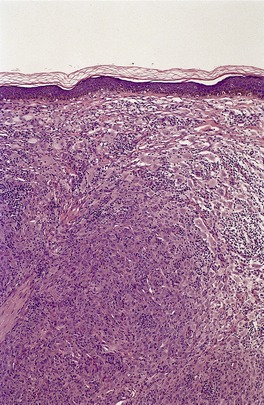
Fig. 40.10
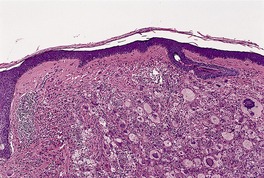
Fig. 40.11
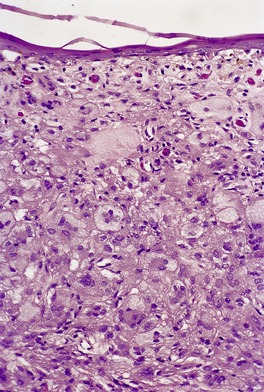
Fig. 40.12
Electron microscopy478.558. and 559.
BENIGN CEPHALIC HISTIOCYTOSIS
Histopathology562
Electron microscopy562
PROGRESSIVE NODULAR HISTIOCYTOSIS
Histopathology450.579. and 583.
XANTHOMA DISSEMINATUM
Histopathology589
Electron microscopy
ERDHEIM–CHESTER DISEASE
Histopathology
SEA-BLUE HISTIOCYTE SYNDROME
Histopathology
GENERALIZED ERUPTIVE HISTIOCYTOMA
Histopathology612. and 613.
PROGRESSIVE MUCINOUS HISTIOCYTOSIS
Histopathology
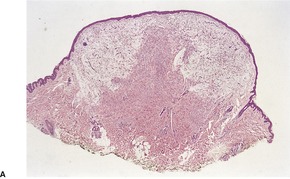
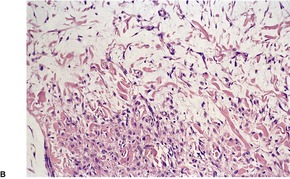
Fig. 40.13
Electron microscopy
MULTICENTRIC RETICULOHISTIOCYTOSIS
Histopathology634.643. and 662.
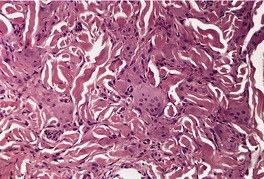
Fig. 40.14
Electron microscopy
RETICULOHISTIOCYTOMA
Histopathology671
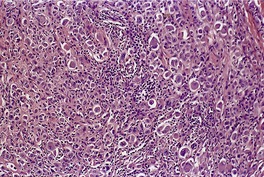
Fig. 40.15
FAMILIAL HISTIOCYTIC DERMATOARTHRITIS
Histopathology
NECROBIOTIC XANTHOGRANULOMA
Histopathology705
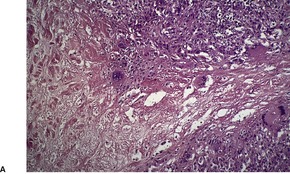
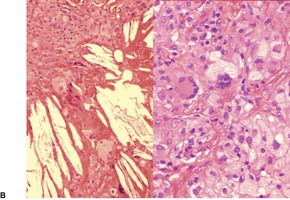
Fig. 40.16
ORBITAL XANTHOGRANULOMA
Histopathology
CUTANEOUS ATYPICAL HISTIOCYTOSIS
Histopathology
Electron microscopy
INDETERMINATE CELL HISTIOCYTOSIS
Histopathology
ROSAI–DORFMAN DISEASE
Histopathology728
GIANT-CELL RICH HISTIOCYTIC DERMATITIS/PANNICULITIS
Histopathology
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
Type
OMIM
Gene locus
Gene
1
267000
9q21.3–q22
Not characterized
2
603553
10q22
PRF1 (encodes perforin)
3
608898
17q25.1
UNC13D (involved in perforin transfer)
4
603552
6q24
STX11 (syntaxin-11)
Histopathology
HISTIOCYTIC SARCOMA
Histopathology
Electron microscopy
REACTIVE HISTIOCYTOSIS
INTRAVASCULAR/INTRALYMPHATIC HISTIOCYTOSIS
Histopathology
XANTHOMATOUS INFILTRATES
Eruptive xanthoma
Tuberous xanthoma
Tendinous xanthoma
Plexiform xanthomatous tumor
Planar xanthoma
Verruciform xanthoma
Papular xanthoma
Facial xanthomatosis
Subcutaneous xanthomatosis
POEMS syndrome
Juvenile xanthogranuloma
Progressive nodular histiocytosis
Necrobiotic xanthogranuloma
Orbital xanthogranuloma
Xanthoma disseminatum
Erdheim–Chester disease
Sea-blue histiocyte syndrome
Tangier disease
Disseminated lipogranulomatosis
Langerhans cell histiocytosis (histiocytosis X)
Congenital self-healing histiocytosis
Lepromatous leprosy
Rhinoscleroma
Malakoplakia
Scars
Arthropod bites
Lymphedema
Dermatofibroma (histiocytoma)
Hamartoma of dermal dendrocytes
Mycosis fungoides
Erythroderma
Etiology and pathogenesis of xanthomas
ERUPTIVE XANTHOMA
Histopathology841
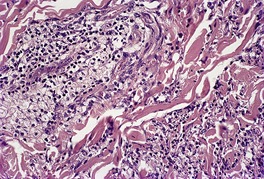
Fig. 40.17
TUBEROUS XANTHOMA
Histopathology
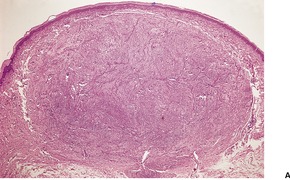
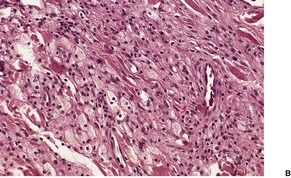
Fig. 40.18
TENDINOUS XANTHOMA
Histopathology
PLEXIFORM XANTHOMATOUS TUMOR
Histopathology858
PLANAR XANTHOMA
Xanthelasma
Intertriginous xanthoma
Xanthoma striatum palmaris
Diffuse (generalized) plane xanthomas
Histopathology
Electron microscopy
VERRUCIFORM XANTHOMA
Histopathology891
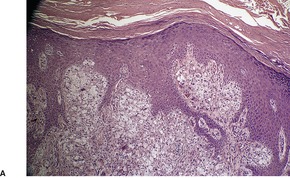
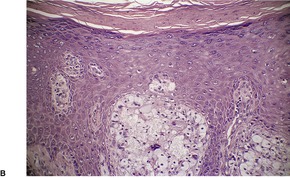
Fig. 40.19
PAPULAR XANTHOMA
Histopathology478. and 935.
Electron microscopy
PAPULAR NEUTROPHILIC XANTHOMA
Histopathology
LANGERHANS CELL INFILTRATES
Langerhans cells
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Cutaneous infiltrates – non-lymphoid
Neutrophil infiltrates938
Histiocytic infiltrates (non-Langerhans cell)951
Juvenile xanthogranuloma953
Benign cephalic histiocytosis954
Progressive nodular histiocytosis955
Xanthoma disseminatum955
Erdheim–Chester disease955
Sea-blue histiocyte syndrome956
Generalized eruptive histiocytoma956
Progressive mucinous histiocytosis956
Multicentric reticulohistiocytosis956
Reticulohistiocytoma957
Familial histiocytic dermatoarthritis958
Necrobiotic xanthogranuloma958
Orbital xanthogranuloma959
Cutaneous atypical histiocytosis959
Indeterminate cell histiocytosis959
Rosai–Dorfman disease959
Giant-cell rich histiocytic dermatitis/panniculitis960
Hemophagocytic lymphohistiocytosis960
Histiocytic sarcoma961
Reactive histiocytosis961
Intravascular/intralymphatic histiocytosis961
The various conditions will be discussed by cell type. For completeness, mention will be made of those conditions fulfilling the definition of ‘cutaneous infiltrates’ but discussed in other chapters. Special histochemical methods and immunoperoxidase techniques using monoclonal antibodies may be required to characterize fully the particular cell which constitutes the cutaneous infiltrate.
Infiltration of the skin by neutrophils is common to numerous diseases of the skin. In most instances the infiltrate is localized to the dermis, although involvement of the epidermis or the subcutaneous fat may occur in various conditions.
Neutrophils (neutrophil polymorphonuclear leukocytes) measure 10–12 µm in diameter in tissue sections. They have two to five distinct nuclear lobes and their cytoplasm contains two distinct types of granules. The larger ones are azurophilic and constitute approximately 25% of the granules in the cytoplasm. They contain myeloperoxidase, bactericidal substrates, cationic proteins, acid hydrolases, and elastase. 1 The smaller granules contain lactoferrin, lysozyme, collagenase, and alkaline phosphatase. 1 These enzymes contribute to the neutrophils’ vital role in the defense against invading microorganisms.
Neutrophils are produced in the bone marrow; their maturation from myeloblasts through various intermediate stages takes approximately 7–10 days. Their production and maturation is under the influence of a specific glycoprotein known as granulocyte colony-stimulating factor.2. and 3. Mature neutrophils are released into the circulation, where they spend approximately 7 hours before entering the tissues. Chemotactic substances guide neutrophils to the site of the infective process or another stimulus. They remain functional in the tissues for 1–2 days. Their demise is poorly understood: they can be phagocytosed by macrophages in the tissues or spleen; some appear to be excreted from mucosal surfaces. 1
The prime function of neutrophils is phagocytosis and the release of the various enzymes stored in the cytoplasmic granules (see above). An unwanted effect is tissue damage caused by some of these enzymes, in particular collagenase and elastase. Although the part played by neutrophils in the phagocytosis and elimination of organisms, immune complexes, and damaged tissue is well understood, their role in dermatoses such as psoriasis, dermatitis herpetiformis, and the neutrophilic dermatoses is not well understood.
All of the conditions characterized by neutrophilic infiltrates in the skin have been discussed in other chapters with the exceptions of Still’s disease and congenital and erosive vesicular dermatosis, both of which may have numerous neutrophils in the inflammatory infiltrate.4. and 5. The various neutrophilic diseases were reviewed in 2006. 6 They are summarized in Table 40.1.
Eosinophils are readily identified in tissues, but their role in the pathogenesis of the various cutaneous diseases in which they are found has been obscure until comparatively recently. 7 It is now known that they are the effector cells for killing helminths and also for causing tissue damage in hypersensitivity diseases. 7 Eosinophils have also been linked to several inflammatory diseases of the skin associated with edema.8.9. and 10. They appear to have a role in the down-regulation of the inflammation associated with hypersensitivity reactions of immediate type. 7 Eosinophils also possess phagocytic activity, but less than that of neutrophils. 7
Eosinophils are polymorphonuclear leukocytes with a bilobed or trilobed nucleus and cytoplasm which contains approximately 20 eosinophilic-staining granules. 11 Ultrastructurally, these granules have an electron-dense core and a relatively radiolucent matrix. 12
The granular core is the site of production of major basic protein, whereas the other granule proteins (eosinophil cationic protein, eosinophil-derived neurotoxin, and eosinophil peroxidase) are found in the matrix. 8 The granule proteins are potent toxins, some of which (major basic protein and eosinophil cationic protein) can directly kill metazoal parasites coated with IgE. Eosinophil cationic protein can also cause local tissue damage when it is released. 13 Major basic protein can cause histamine release from basophils, and most of the granule proteins can induce a wheal-and-flare reaction. 8 Major basic protein can be detected in the tissues in atopic dermatitis14 and in some cases of urticaria, 12 even in the absence of significant tissue eosinophilia. 8 This has important pathogenetic implications for these two diseases.
Other substances produced by the eosinophil include Charcot–Leyden crystal protein, which has lysophospholipase activity, arylsulfatase, leukotriene C4, and platelet-activating factor. 8 Thromboembolic disorders are more common in patients with the hypereosinophilic syndrome than in other people, presumably as a consequence of enhanced production of platelet-activating factor. 15
Eosinophils originate in the bone marrow, where they spend 3–6 days before being released into the circulation. 11 Several factors such as granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and interleukin-5 (IL-5) stimulate the production of eosinophils.16. and 17. They are in the blood for a short time and then enter the tissues. 11 Only a small proportion of the total number of eosinophils are circulating at any time. Eosinophils appear to go through a late differentiation stage after they have entered the bloodstream.15. and 18. Some eosinophils develop low-density cytoplasm (‘hypodense eosinophils’) which corresponds with activation of the cell.15. and 18. Chemotaxis for eosinophils is mediated by GM-CSF, IL-5, leukotriene B4, platelet activation factor, and complement fraction 5. 16 Of particular importance is the role of IL-5, which has selective specificity for eosinophils.16.17. and 19. The gene for IL-5 is located at 5q31.1. 17 The selectivity of the eosinophil response to a particular stimulus is due to the receptor profile expressed by eosinophils, which is predominantly the CCR3 receptor. 17 Genetic mutations involving the platelet-derived growth factor receptor genes have been pathogenetically linked to clonal eosinophilia. 20
Eosinophils are a conspicuous component of the inflammatory cell infiltrate in a number of inflammatory and neoplastic disorders. They are listed in Table 40.2.
The distribution of the eosinophils within the skin may be characteristic. This aspect is covered in the discussion of the various entities in other sections of the book. One pattern of distribution of eosinophils within the dermis – interstitial eosinophils – is characteristic of certain diseases. The term ‘interstitial eosinophils’ refers to the presence of eosinophils between collagen bundles in the intervascular dermis. Eosinophils are invariably present in a perivascular location as well. Interstitial eosinophils are characteristic of various parasitic infestations, particularly arthropod bites (p. 659), but they are also found in certain drug reactions, including the drug hypersensitivity syndrome21 (see Ch. 20), urticaria (p. 206), PUPPP (toxic erythema of pregnancy; p. 227), Wells’ syndrome (see below), the urticarial stage of bullous pemphigoid (p. 156), eosinophilic, polymorphic and pruritic eruption of radiotherapy (see p. 528), the hypereosinophilic syndrome (p. 941), and dermal hypersensitivity reaction (see below).
A discussion of Wells’ syndrome, the hypereosinophilic syndrome, pachydermatous eosinophilic dermatitis, dermal hypersensitivity reaction, and eosinophilic pustulosis follows. Mention will be made here of the condition known as dermatitis cruris pustulosa et atrophicans. 22 It is a frequent but poorly understood tropical skin condition with intraepidermal pustules and a dermal infiltrate of neutrophils and eosinophils forming flame figures. 22
Wells’ syndrome (eosinophilic cellulitis)23.24.25.26. and 27. is a disorder of unknown pathogenesis characterized by the tissue reaction pattern known as ‘eosinophilic cellulitis with flame figures’ (see p. 15).
Clinically, there are edematous infiltrated plaques resembling cellulitis, often with blister formation. 28 This is followed by the development of slate-gray morphea-like induration which resolves, usually without trace, over 4–8 weeks.24.29. and 30. Recurrent lesions may develop over a period of months to years. A papulonodular form is uncommon.31. and 32. Milder cases have annular or circinate erythematous plaques. The condition known as ‘eosinophilic annular erythema’ may not be a variant of Wells’ syndrome as once thought. 33 Subcutaneous nodules are extremely rare. 34 Insect bite-like lesions may occur. 35 There is a predilection for the extremities and trunk; rarely, the face is involved as a major clinical feature. 36 The lesions followed the lines of Blaschko in one case. 37
Wells’ syndrome may occur at any age, although onset in childhood is uncommon.30.34.38.39.40.41.42. and 43. Several cases occurring in the same family have been documented.44. and 45. In one family, the skin lesions were associated with a dysmorphic habitus, learning disability, and elevated plasma IL-5. 45
Although most cases of Wells’ syndrome are idiopathic, some are associated with arthropod bites, parvovirus B19 infection, 46 parasitic infestation (such as giardiasis and toxocariasis), drug allergy, tetanus vaccine,47. and 48. thiomersal-containing vaccines, 49 or an atopic history.24.26.50.51.52.53. and 54. It has followed varicella infection in a child41 and been associated with HIV infection, 55 molluscum contagiosum, 56 eosinophilic fasciitis, 57 the Churg–Strauss syndrome, 58 bronchogenic carcinoma, 59 ulcerative colitis, 60 and the hypereosinophilic syndrome.61.62. and 63. It has also followed the use of 2-chlorodeoxyadenosine in the treatment of hairy cell leukemia. 64
It has been suggested that circulating CD4+ CD7− T cells play a pivotal role by producing IL-5.57. and 65. Serum and tissue levels of this cytokine appear to correlate with clinical activity. 16
Many cases resolve spontaneously, but systemic corticosteroids are often used to treat this condition. Minocycline and doxycycline have also been used in isolated cases. 66 Interferon (IFN)-α was used in a case with a clonal population of T cells producing IL-5. 57
In early lesions of Wells’ syndrome there is dermal edema and massive infiltration of eosinophils, both interstitial and angiocentric. Subepidermal blisters containing eosinophils may form. 28 Blisters may also form from spongiotic vesiculation. 69 After 1 week scattered histiocytes and characteristic ‘flame figures’ are found. The flame figures are surrounded, in part, by a palisade of histiocytes and a few multinucleate giant cells. Uncommonly, there are numerous multinucleate giant cells present. 70
The inflammatory process involves the entire thickness of the dermis, and often the subcutis as well. Localization to the subcutis has been reported as eosinophilic panniculitis (see p. 475). Extensive necrotizing granulomas have been reported in the subcutis in this condition. 39 Rarely, the inflammatory infiltrate extends into the fascia and muscle. 71
The flame figures consist of eosinophil granule major basic protein encrusted on otherwise normal collagen. 29 There is no mucopolysaccharide or lipid, but sometimes basophilic fibrillar material may be seen at the periphery of the eosinophilic material (Fig. 40.1). There is a superficial resemblance to the Splendore–Hoeppli phenomenon, which may develop around metazoal parasites in the tissues.
Wells’ syndrome. (A) A flame figure is shown. (B) There are many eosinophils in the surrounding dermis. (H & E)
The tissue reaction pattern of eosinophilic cellulitis with flame figures may be seen in a number of disparate conditions,23.67. and 72. including arthropod reactions,67. and 73. other parasitic infestations, 74 internal cancers, 75 etanercept injection sites, 76 dermatophyte infections, 23 bullous pemphigoid, 23 herpes gestationis, 23 allergic eczemas, 23 and eosinophilic ulcer of the oral mucosa.77.78. and 79. It is uncommon in all of these conditions, except eosinophilic ulcer of the oral mucosa. Atypical CD30+ cells have also been reported in the cellular infiltrate in this condition. 80 This reaction pattern has also been reported in association with eosinophilic folliculitis, as a manifestation of a drug reaction. 81 The clinical setting and other histopathological features allow the conditions listed above to be distinguished from Wells’ syndrome.
In eosinophilic annular erythema, in addition to the features seen in Wells’ syndrome there may be basal vacuolar change and some mucin deposition in the dermis. 33
Free eosinophil granules are found coating collagen bundles in the flame figures, but the collagen bundles are not damaged. 68 Numerous intact eosinophils are also present in the adjacent dermis.
The hypereosinophilic syndrome (OMIM 607685) is a systemic disorder with involvement of one or more organs and persistent hypereosinophilia (>1.5 × 109/L) in the absence of any identifiable cause.82. and 83. Recent work suggests that a PDGFRA/FIP1L1 fusion gene may be responsible in some cases (see below). It encompasses a spectrum of disorders which includes eosinophilic leukemia and Löffler’s syndrome. Eosinophilic leukemia may initially present as a hypereosinophilic syndrome. 84 Cardiac involvement, which may be fatal, is quite common; the lungs, skin, liver, and central nervous system may also be involved.
Cutaneous lesions, which take the form of pruritic erythematous papules and nodules or urticaria and angioedema, are present in one-half of cases. 85 Rarely they are the only manifestation of the syndrome.86. and 87. Other mucocutaneous presentations include mucosal ulcerations, 88 erythema annulare centrifugum, 89 purpuric lesions, eosinophilic cellulitis, 63 skin necrosis,90. and 91. superficial venous thrombophlebitis, 92 thromboangiitis obliterans, 93 erythroderma, 94 and livedo reticularis. 95 Elevated levels of IgE, IL-2, IL-5, IL-10, and soluble IL-2 receptor may be present. 96 Dermal endothelial cells express eotaxin, a chemokine that is a potent chemoattractant for eosinophils. 97
Cases presenting with episodic or transient angioedema and eosinophilia, but without involvement of other organs, are regarded as a separate entity with a benign clinical course.10.82. and 98. Likewise, the syndrome of hyperimmunoglobulinemia-E with recurrent staphylococcal skin infections, defective neutrophil chemotaxis, and peripheral eosinophilia is a distinct entity.99.100.101.102. and 103. There are two distinct hypereosinophilic syndromes: the better known one, Job’s syndrome (OMIM 147060), is autosomal dominant and caused by a mutation in the STAT3 gene located at 4q21; the other one is an autosomal recessive form (OMIM 243700), in which the mutation has not been characterized. Early-onset eczematous lesions, often misdiagnosed as atopic dermatitis, and characteristic facies are other manifestations of Job’s syndrome.101. and 104. Rituximab, an antibody against CD20, has been used to treat a case. 105 The case that presented with nodular eosinophilic infiltration of the skin and immunoglobulin isotype imbalance is probably a separate condition. 106
The precise origin of the hypereosinophilic syndrome is unknown, but it appears to involve a dysregulation of interleukins-3 and -5 (IL-3 and -5) and/or granulocyte–macrophage colony-stimulating factor. 20 Recently, the FIP1L1-PDGFRA (F/P) fusion gene has been identified in some patients with hypereosinophilic syndrome.107. and 108. Such cases have overlap features with chronic eosinophilic leukemia. The two genes that fuse are closely associated at gene map locus 4q12.
Mepolizumab, an antibody to IL-5, appears to be a promising targeted therapy for the hypereosinophilic syndrome. 20 Systemic corticosteroids are the current mainstay of treatment. 109 Infliximab has also been used. 110
There is a variable superficial and deep, predominantly perivascular, infiltrate of eosinophils, with variable numbers of lymphocytes, plasma cells, and mast cells. 112 Dermal edema is present in urticarial lesions. Thrombosis of small vessels in the dermis is a rare finding. 95 The microthrombi often contain eosinophils. 109
In the syndrome of recurrent angioedema with eosinophilia the infiltrate is primarily mononuclear, with only a few eosinophils. 113 However, with immunofluorescence there is extracellular localization of eosinophil major basic protein.
Pachydermatous eosinophilic dermatitis is possibly a variant of the hypereosinophilic syndrome. It is associated with a generalized pruritic papular eruption arising on a pachydermatous base, hypertrophic lesions in the genital area, and peripheral blood eosinophilia. 114 The reported cases were in South African black teenage girls. The etiology is unknown.
The lesions showed an eosinophil-rich lymphohistiocytic infiltrate and variable fibrosis of the dermis. The infiltrate varied in amount and distribution. Interstitial eosinophils were often present and there was abundant eosinophil granule major basic protein. 114
Dermal hypersensitivity reaction (DHR) is the most controversial concept in dermatopathology, because of its inconsistent usage, its variable clinicopathological correlations, its enigmatic nature, and the emotive climate that accompanies the use of the term.115. and 116. LeBoit has termed it ‘the last refuge of scoundrels’. 117 Against this background the author of this book now believes that after practicing dermatopathology for 35 years, no other concept/diagnosis appropriately encompasses the pathological findings that occur in some patients in a defined etiological setting. The author is also aware of the disdain held for converts by some persons: ‘the impudence of a bawd is as nothing to that of a convert’ – George Saville, Lord Halifax (1633–1695). The term used by Kossard and colleagues, urticarial dermatitis, comes closest to an appropriate substitute designation for DHR, but by their own admission, urticarial dermatitis applies only to a subset of patients having DHR. 118 Some of the cases reported as papular dermatitis (subacute prurigo, ‘itchy red bump’ disease) are probably further examples of DHR.119. and 120.
The clinical presentation is variable, but lesions are usually intensely pruritic. There may be persistent urticarial plaques, urticated erythema, or a papular eruption involving the trunk and limbs. The latter presentation is usually symmetrical. Involvement of the face and neck is much less common. It may occur at any age. Lesions persist for months or years. Resolution of the lesions, with subsequent recurrence, is sometimes seen.
Dermal hypersensitivity reaction may be idiopathic or follow well-documented events such as drug ingestion, vaccination, an arthropod or snake bite, infection, particularly of viral type, and an internal malignancy, including lymphoma.116. and 121. The author has seen many cases with a generalized eruption that followed local skin contact with the sap of the papaya or the macadamia nut tree. The generalized rash was not an autoeczematization.
Treatment with mycophenolate mofetil, 122 or low doses of prednisone combined with either dapsone or hydroxyurea has been used. 123
The most common pattern is a superficial and, to a much less extent, deep perivascular infiltrate of lymphocytes admixed with a few eosinophils, combined with many interstitial eosinophils (Fig. 40.2). They are sometimes more numerous than in arthropod bite reactions, which can usually be excluded because of the symmetrical nature of the rash or the presence of large plaques. It should be remembered that not every lesion in a bite reaction, particularly scabies, is due to arthropod contact. Some lesions are examples of a hypersensitivity reaction.
(A) Dermal hypersensitivity reaction. (B) There is a tight perivascular infiltrate of lymphocytes and fewer eosinophils than usual. (H & E)
Mild urticarial edema and mild epidermal spongiosis, which is usually more diffuse than seen with arthropods, are often present. Epidermal changes of scratching are present in older lesions. A few basal apoptotic keratinocytes are sometimes present.
Another pattern seen in DHR resembles a lymphocytic vasculitis, with a tight perivascular cuff of lymphocytes, with a few admixed eosinophils. Fibrin and red cell extravasation are rarely present, suggesting that the infiltrate may be directed at antigen-processing endothelial cells, rather than representing a true vasculitis. 124
Eosinophilic pustulosis is an appropriate designation for a spectrum of cases originally reported as eosinophilic pustular folliculitis of infancy (see p. 403). 125 Although it was originally regarded as a dermatosis of the scalp, cases with lesions in other sites have been reported. Furthermore, this condition was originally regarded as a follicular process similar to Ofuji’s disease but cases with a predominantly interfollicular infiltrate have since been reported. The papulopustular lesions reported in the genital region appear to belong to this spectrum. 126
The etiology is unknown. Scabies infection has not been present.
There is a heavy dermal infiltrate of lymphocytes, neutrophils, and numerous eosinophils. By definition, there is no primary folliculitis. There is usually no subepidermal edema, and the infiltrate is more polymorphic than in Wells’ syndrome.
Plasma cells are not usually present in the peripheral blood or normal skin, although they may be found in normal mucous membranes. However, they may be a component of the inflammatory infiltrate in a wide range of dermatoses and tumors of the skin. 127 These conditions will be considered after a discussion of the normal plasma cell.
Plasma cells are terminally differentiated cells which are derived from antigenically stimulated B lymphocytes. 127 During their lifespan of 2–3 days they continuously synthesize and secrete antibodies that have specificity for the particular antigen that stimulated the plasma cell precursor to proliferate and differentiate.
Plasma cells have abundant basophilic cytoplasm and an eccentrically placed nucleus with coarse chromatin granules, which are often distributed in a cartwheel pattern. Occasionally the cytoplasm contains a round eosinophilic inclusion that may displace the nucleus to the periphery or be liberated into the stroma. These Russell bodies, which may measure up to 20 µm in diameter, result from the accumulation of immunoglobulins and glycoproteins in the cytoplasm. 128 They are PAS positive and diastase resistant. Russell bodies are particularly prominent in the inflammatory infiltrate in rhinoscleroma.
Ultrastructural examination reveals a well-developed rough endoplasmic reticulum with numerous ribosomes. These are involved in the synthesis of a particular immunoglobulin. The endoplasmic reticulum is the site of formation of the Russell bodies. Crystalloid inclusions, iron, and even bacteria have been found in the cytoplasm of plasma cells in various circumstances. 127
Numerous basophilic extracellular bodies, reminiscent of yeast cells, have been found in the dermis in association with plasma cell infiltrates. 129 These structures, known as plasma cell bodies, may measure up to 5 µm in diameter and are derived from the cytoplasm of plasma cells. 129
In contrast to B lymphocytes, plasma cells have very small amounts of immunoglobulin on their cell membrane, although it may be demonstrated in the cytoplasm by immunoperoxidase methods. This may be useful in distinguishing reactive proliferations of plasma cells, which are polyclonal, from plasmacytomas and myelomatous infiltrates, which are monoclonal. Plasma cells stain for CD79a; CD38 is used as a marker in flow cytometry. CD138 is another marker for plasma cells.
Plasma cells may be a prominent component of the inflammatory infiltrate in a number of dermatoses and neoplastic disorders. 127 It should be remembered that plasma cells are almost invariably present in any condition involving the lips and other mucous membranes. 130 For some inexplicable reason they may also be prominent in lesions of the forehead and scalp, particularly in keratoses and skin cancers previously subjected to cryotherapy. 131
Plasma cells are particularly prominent in the inflammatory infiltrate in syphilis (p. 574), granuloma inguinale (p. 567), chancroid (p. 568), yaws (p. 577), rhinoscleroma (p. 568), erythema nodosum leprosum (p. 566), necrobiosis lipoidica (p. 181), the nodular form of primary cutaneous amyloidosis (p. 381), chronic folliculitis (p. 404), Kaposi’s sarcoma (p. 913), and syringocystadenoma papilliferum (p. 780). 127 They may be prominent at the lower edge of the dermal infiltrate in a subgroup of patients with mycosis fungoides (p. 976), 132 and at the periphery of the rare entity known as inflammatory pseudotumor. This latter lesion has the low-power appearance of a lymph node with variable central fibrosis, but there are no sinuses present.133.134. and 135.
Plasma cells are a less consistent feature in certain deep fungal infections, cutaneous lupus erythematosus, rosacea, scleroderma, pseudolymphoma, persistent light reactions, in drug reactions, 136 and the reaction to certain arthropods. 131 A panniculitis rich in plasma cells may be seen in scleroderma, morphea profunda, dermatomyositis, and Sjögren’s syndrome. Rarely, they are present in lichen planus. 137 Plasma cells are a diagnostic feature of one variant of Castleman’s disease (p. 946). Plasma cells are often present in the stroma of various tumors such as squamous cell carcinomas and basal cell carcinomas, and malignant melanomas, particularly if ulceration has occurred.
These conditions should probably have been considered in Chapter 41 with the lymphoid infiltrates. To do so would have created certain logistical problems. For this reason they are again considered in this chapter. Cutaneous plasmacytomas are rare monoclonal proliferations of plasma cells which are usually associated with underlying multiple myeloma, extramedullary (soft tissue) plasmacytomas or, rarely, plasma cell leukemia.138. and 139. These secondary cutaneous plasmacytomas are usually multiple. They may arise by direct spread from an underlying tumor deposit in bone or soft tissue, or by metastatic spread via lymphatics or blood vessels. 140 Cutaneous plasmacytomas develop in only a small percentage of cases of multiple myeloma, and of extramedullary plasmacytoma.138.140.141.142. and 143. Only 20 cases were noted in a series of 2357 patients with the diagnosis of multiple myeloma. 144 They are a bad prognostic sign.140. and 145. Sometimes cutaneous lesions are the presenting sign of multiple myeloma and, rarely, they may antedate the development of the full-blown disease. 146 It has been suggested that IgA- and IgD-producing plasmacytomas are disproportionately represented in the skin. 147
Primary cutaneous plasmacytomas, which by definition arise without concomitant bone marrow or soft tissue disease, are exceedingly rare.148.149.150.151.152. and 153. They are included in the current WHO/EORTC classification with the B-cell lymphomas (see pp. 972 and 998). They constitute only 2–4% of all cases of primary extramedullary plasmacytomas. 154 They usually grow slowly and are solitary; multiple lesions are sometimes present.155.156.157.158. and 159. The prognosis is not as good as previously thought: visceral metastases and death occur in over one-third of cases.160. and 161. Transformation of a cutaneous plasmacytoma into a CD30+ diffuse large B-cell lymphoma has been reported, as a consequence of Epstein–Barr virus infection. 162 Rarely a local triggering stimulus may be involved in the development of a primary cutaneous plasmacytoma. One such case involved recurrent herpes simplex virus (HSV-1) infection of the lip with the development of a plasmacytoma at the site after 15 years of recurrent infection. 154 The authors hypothesized that recurrent infection resulted in the toll-like receptor activation and, in turn, interleukin-6 release producing plasma cell proliferation, transformation, and survival. 154 Cases associated with transplantation and chronic immunosuppression have been reported.159. and 163.
Plasmacytomas are dusky red or violaceous dome-shaped nodules which measure 1–5 cm in diameter. They have a predilection for the trunk, but they may also develop on the extremities and the face.
Multiple myeloma (myelomatosis) is a malignant proliferation of plasma cells that typically involves the bone marrow but may also involve other tissues. 164 It is a disease of older persons; the median age in a large series from the Mayo Clinic was 66 years. 165 The median duration of survival was 33 months. 165 Myeloma usually presents with bone pain, anemia, renal insufficiency, hypercalcemia, and proteinuria. 164 A monoclonal spike is found in the electrophoretic pattern of the serum, and sometimes of urine, in which Bence Jones protein may also be detected. Amyloidosis may subsequently develop. Cutaneous manifestations of multiple myeloma include the development of xanthomas, amyloid deposits, non-specific erythemas, neoplastic plasma cell infiltrates, 166 pyoderma gangrenosum-like lesions, cutaneous infections, including herpes zoster, and hemorrhagic lesions.164.167. and 168. Other diseases can occur following autologous peripheral stem cell transplants and various types of bone marrow transplantation used in the treatment of myelomatosis. 169
Cutaneous plasmacytomas are circumscribed non-encapsulated dense infiltrates of plasma cells, situated usually in the reticular dermis but sometimes involving the subcutis as well. The epidermis is often stretched over the deposit, but ulceration is uncommon. The plasma cells show variable maturation (Fig. 40.3). Russell bodies are often present in lesions with many mature plasma cells. In secondary and rapidly growing primary lesions there is more variation in the size and the maturation of the plasma cells, with some binucleate cells and scattered mitoses. 170 Rare variants of plasma cell tumors include pleomorphic, blastic, signet-ring, small cell, clear cell, and spindle-cell forms. 171 Sometimes the cells are immature and resemble immunoblasts or the cells of a malignant lymphoma. These cases with dedifferentiated cells are now classified as lymphoplasmacytoid lymphomas or immunocytomas. 172 Their monoclonal nature can be confirmed by immunohistochemistry. Occasional plasmacytomas are polyclonal. 173 The specific plasma cell antibody PC-1, and CD79a can be used for their confirmation. 127 Plasmacytomas are often negative for CD45 and can be cytokeratin positive, leading to an erroneous interpretation of cancer. The cells do not usually express B-lineage cell surface markers such as L26 (CD20). 161 Cutaneous plasmacytomas express CD117 in a cytoplasmic, or membranous and cytoplasmic distribution, with varying degrees of staining intensity. 174 The methyl green–pyronin stain, in which the cytoplasm of plasma cells stains red, may be used to confirm the cell type in less well-differentiated deposits. 175 The expression of syndecan-1 (CD138) is a sensitive marker for cutaneous plasmacytomas, independent of cytological differentiation.144. and 176. It is not always expressed in myeloma deposits. 177
Plasmacytoma composed of mature plasma cells. (H & E)
There is one report of needle-like crystalloid inclusions, possibly representing phagocytosed protein, in the cytoplasm of macrophages that were admixed with the plasma cells. 178 Further reports document patients with myeloma with crystalline deposits in the skin, but without accompanying plasma cells.179. and 180. The term crystal-storing histiocytosis has been used for these cases. 180 Amyloid is another uncommon finding in the cutaneous lesions.
The immature plasma cells found in some rapidly growing plasmacytomas and cutaneous deposits of multiple myeloma have less abundant rough endoplasmic reticulum than do mature cells. 160
A heterogeneous group of cutaneous diseases have been associated with multiple myeloma or with a monoclonal gammopathy. These include necrobiotic xanthogranuloma, xanthoma disseminatum, generalized plane xanthomas, lichen myxedematosus and scleromyxedema, scleredema, erythema elevatum diutinum, subcorneal pustular dermatosis, pyoderma gangrenosum, dermatitis herpetiformis, a subepidermal bullous dermatosis, 181 acquired angioedema, angioimmunoblastic lymphadenopathy, cutaneous T-cell lymphoma, mycosis fungoides, and the Sézary syndrome. 182
A monoclonal protein is present in approximately 75% of cases of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein and skin lesions).183.184.185. and 186. It is also known as Crow–Fukase syndrome. The cutaneous lesions include hyperpigmentation, hypertrichosis and skin thickening. 187 Glomeruloid hemangiomas also occur (see p. 900). Xanthoma cells have been described in the hyperpigmented patches of one patient. 188 Increased serum levels of vascular endothelial growth factor have been reported in this syndrome. 189 Increased serum levels of IL-6, and also HHV-8 infection have been reported, as also occurs in Castleman’s disease (see below). 190 Improvement of the skin lesions in POEMS syndrome has been reported after UVA-1 phototherapy. 191
In Schnitzler’s syndrome there is chronic urticaria associated with macroglobulinemia, but criteria for the diagnosis of Waldenström’s disease (see below) are lacking. 192
Waldenström’s macroglobulinemia is a lymphoproliferative disorder of the elderly in which IgM-producing lymphoplasmacytoid cells proliferate in the bone marrow and/or lymph nodes and spleen.193. and 194. It is now regarded as a lymphoma of small B lymphocytes, plasma cells, and plasmacytoid cells (see p. 997). Clinical features include weight loss, weakness, anemia, and a bleeding diathesis.
Cutaneous manifestations are usually non-specific and result from hyperviscosity of the blood and the bleeding tendency. They include purpura, discoloration of the fingertips and toes, leg ulcers, 195 urticaria, bullae, and angioedema.196. and 197. Specific skin lesions are quite rare. They take the form of translucent papules composed of deposits of monoclonal IgM (macroglobulinosis cutis),198. and 199. and of violaceous plaques, nodules, or macular lesions on the face, trunk, or proximal parts of the extremities; the plaques, nodules, and macules are composed of lymphoplasmacytoid cells.147.194.198.200. and 201. The cutaneous lesions usually develop later in the course of the disease, although they may be the presenting feature. 200
The rare translucent papules consist of eosinophilic hyaline deposits filling the papillary and upper reticular dermis. Artifactual clefts may be present. 198 Sometimes the material encases the hair follicles; it may undergo transepithelial elimination. The hyaline deposits are strongly PAS positive, but negative with stains for amyloid. They are monoclonal for IgM, using immunofluorescent techniques.
The plaques and tumors are composed of a dense infiltrate of lymphoplasmacytoid cells in the reticular dermis. 193 Occasional binucleate cells and mitotic figures are present. Some of the cells contain intranuclear inclusions which are PAS positive. 147 Hyaline material, which is monoclonal IgM, is sometimes present between the cells. IgM has also been reported along the basement membrane zone of both lesional and non-lesional skin.202. and 203. Sometimes a leukocytoclastic vasculitis is also present. 197
The hyaline deposits are composed of granular material which is electron dense. Fibrils are usually absent,198. and 199. although they were present in one case. 204 The lymphoplasmacytoid cells in the infiltrative lesions have abundant ribosomes, and sometimes intranuclear inclusions composed of granular material.
‘Systemic plasmacytosis’ has been applied in cases with multiple brownish plaques, asymptomatic generalized lymphadenopathy and polyclonal hypergammaglobulinemia.205.206. and 207. There is one report of a patient with systemic plasmacytosis later developing a nodal T-cell lymphoma. 208 The term ‘cutaneous plasmacytosis’ has been used for the skin lesions when lymphadenopathy is absent.206.209.210.211.212. and 213. This distinction is somewhat artificial as some cases of cutaneous plasmacytosis have subsequently developed systemic disease.214. and 215. The term ‘cutaneous and systemic plasmacytosis’ is now being used for this entity, reflecting the variable systemic involvement that may accompany cutaneous disease. 216
Cutaneous plasmacytosis is a rare skin disorder, most common in Japan, characterized by multiple dark-brown papules and plaques, located predominantly on the trunk.217. and 218. There is often a polyclonal hypergammaglobulinemia. Some cases are associated with an underlying malignancy. 217 HHV-8 was not detected in one study. 219 A case has been reported in a child. 220
Interleukin-6 (IL-6), a major plasma cell growth factor, has been increased in the serum in some cases. 221
Cutaneous plasmacytosis is characterized by moderately dense, superficial and deep, perivascular infiltrates in the dermis, composed of mature plasma cells and a small number of lymphocytes. 205 Sometimes lymphoid aggregates and a few multinucleated giant cells are present. A few interstitial histiocytes are usually seen. 219 There is some resemblance to the lesions of secondary syphilis. There is no light chain restriction. 219
An almost endless variety of terms has been used for comparable lesions involving the penis (plasma cell erythroplasia, balanitis circumscripta plasmacellularis, Zoon’s balanitis), vulva (vulvitis circumscripta plasmacellularis, Zoon’s vulvitis), lips (plasma cell cheilitis), 222 and other mucosal surfaces (plasma cell orificial mucositis,223.224. and 225. atypical gingivostomatitis, 226 plasmocytosis circumorificialis227). The term ‘plasmacytosis mucosae’ has the advantages of relative simplicity, of applying to all mucosal sites, and of indicating that plasma cells are an important component of the inflammatory infiltrate. Oral and genital involvement have been reported in the same patient. 228
Plasmacytosis mucosae is a rare disorder which most frequently involves the glans penis and/or prepuce of older uncircumcised men (Zoon’s balanitis).227.229.230.231.232.233. and 234. It has also been reported in circumcised males. 235 Vulval lesions (Zoon’s vulvitis), which usually involve adults, are exceedingly rare, with fewer than 50 cases reported.229.236.237.238.239.240. and 241. It may be the cause of intractable vulvar pruritus. 242 Genital lesions usually take the form of a solitary, asymptomatic, sharply defined red-brown glistening patch measuring 1–3 cm in diameter. Occasionally, several lesions may be present. A tumorous variant has also been described (plasmoacanthoma). 223 A plasmoacanthoma in the perianal region has been reported in a patient with intertriginous plasmacytosis. 243
The lesions are usually resistant to treatment, although circumcision has resulted in the disappearance of some lesions of the glans. 232 The etiology is unknown, 236 although herpes simplex antigen has been detected in rare cases, 244 and one case has been associated with autoimmune polyglandular endocrine failure, raising the possibility of autoimmunity in the etiology. 245 It is most probably a chronic, reactive, principally irritant mucositis. 246
Circumcision, as mentioned, is the usual treatment for Zoon’s balanitis, but success with topical steroids and laser have been reported. 247 Tacrolimus (topical) has also been used with a successful outcome.248.249. and 250. Two cases of Zoon’s balanitis have been treated successfully with pimecrolimus 1% cream. 251
There is a dense, often band-like infiltrate of inflammatory cells in the upper dermis, which may extend to the level of the mid-reticular dermis. The infiltrate is composed predominantly of polyclonal plasma cells,223.224. and 233. sometimes containing Russell bodies. The number of plasma cells seems to vary with the stage of the disease; sometimes they are sparse. 252 There may also be lymphocytes, mast cells, occasional eosinophils and even neutrophils in the infiltrate. Neutrophils are more common beneath the epidermis. 234 The presence of lymphoid follicles is a rare event.
Blood vessels are prominent, with an increase in number and some dilatation. There is often extravasation of erythrocytes. Deposits of hemosiderin may be present, although they have not been a feature of non-genital lesions. With time, there is some fibrosis. Weyers and colleagues, in a study of 45 cases, stated that fibrosis, extravasated erythrocytes, siderophages, atrophy of the epidermis, and a dense infiltrate with predominance of plasma cells seemed to reflect more advanced lesions. 234
The overlying epidermis is usually attenuated; frank ulceration occurs in only a minority of cases (Fig. 40.4). In early lesions it may be of normal thickness with focal parakeratosis. The epidermis is often mildly edematous (spongiotic) and may contain a few inflammatory cells and erythrocytes. Mucinous metaplasia has been reported in Zoon’s vulvitis. 253
Zoon’s balanitis. (A) Ulceration is present. (B) There is increased vascularity and many plasma cells. (C) Plasma cells are present beneath a mildly attenuated epidermis. (H & E)
Sometimes, cases diagnosed initially as Zoon’s balanitis turn out to be examples of other diseases, such as psoriasis, lichen planus, or allergic contact dermatitis. 234
In plasmoacanthoma there is an acanthotic and hyperkeratotic epidermis overlying a polypoid tumor composed of a dense dermal infiltrate of mature plasma cells. 243 Russell bodies are also present.
The plasma cells in the infiltrate contain considerable rough endoplasmic reticulum. Phagolysosomes may be present. There are also macrophages containing siderosomes.
Castleman’s disease (giant lymph node hyperplasia) is a distinctive lymphoid hyperplasia that occurs predominantly in the mediastinum of young to middle-aged persons. 254 There are two histological variants, a common hyaline vascular type and a less common plasma cell type, which often has systemic manifestations. A mixed variant, the intermediate type, also occurs. In addition, two clinical types have been described: localized disease and multicentric Castleman’s disease. 255
Cutaneous manifestations of multicentric Castleman’s disease are usually non-specific. They include plane xanthomas and vasculitis. 256 Erythroderma has been described. 257 Cutaneous involvement is rare, with only a few cases reported.254.258. and 259. Cutaneous plasmacytosis has also been reported in patients with multicentric Castleman’s disease.190. and 260. An association with POEMS syndrome (see above) has been recorded. 261 It may represent a variant of Castleman’s disease.
A variant of the hyaline vascular type with lesions confined to the subcutis and skeletal muscle has been reported. 262 The cases were not associated with POEMS syndrome, human herpesvirus type 8, or monoclonal rearrangements of IgH. 262 The lesions ranged in size from 4 to 6 cm in diameter. They had a yellow-brown to red-brown color. 262
The pathogenesis is unknown, but elevated levels of interleukin-6, a cytokine necessary for the maturation of B lymphocytes into plasma cells, are often present. Interleukin-6 also stimulates endothelial hyperplasia. 263 Human herpesvirus type 8 (HHV-8) has been implicated in the etiology of multicentric Castleman’s disease.264. and 265. The cells are usually polyclonal in origin, but rare cases with a monoclonal population of cells have been noted. 255 A sarcoma of presumptive follicular dendritic cell origin has been reported in a case of hyaline–vascular Castleman’s disease of the skin and subcutis; 266 in another case a clonal cytogenic aberration involving the long arm of chromosome 12, with rearrangement of the high-mobility group protein I-C (HMGIC) gene, resulted in a clonal proliferation of follicular dendritic cells. 267 A recent report has suggested that HIV-positive multicentric Castleman’s disease may arise from extrafollicular B cells. 268
Localized Castleman’s disease is generally curable by surgical excision, whereas the multicentric variant often requires systemic therapy and has a poorer prognosis. 255
Cutaneous cases are so rare that few histological reports exist. Reported cases have had a circumscribed nodule composed of ill-defined lymphoid follicles with a mantle of small lymphocytes giving an ‘onion-skin’ appearance. These mantles were traversed by hyalinized capillary-sized vessels. The interfollicular zones were composed of lymphocytes, and plasma cells of polyclonal type. 254 Another case was described as showing a deep dermal and subcutaneous nodular infiltrate of plasmacytoid cells without atypia, and an increased vascular proliferation. 259
Mild to marked follicular dendritic cell dysplasia may be present in cases localized to the subcutis and muscle. 262 The cells are highlighted by CD21 and CD35 stains.
Two cases of the apparently unique condition of angioplasmocellular hyperplasia have been reported. Lesions are composed of a proliferation of small blood vessels, some with vacuolated cytoplasm, and a mixed inflammatory cell infiltrate with a predominance of polyclonal plasma cells. 269 The lesions presented as solitary nodules on the trunk. The author has also seen a case.
Although an increase in mast cells is seen in a variety of inflammatory dermatoses and some tumors, the presence of large numbers of these cells is almost confined to mastocytosis. Aspects of the normal mast cell will be considered before describing the pathological conditions.
Mast cells, which measure 8–15 µm in diameter, are round, oval, or fusiform in shape and have a central nucleus and cytoplasm which contains lightly basophilic granules. 270 The granules stain metachromatically with the toluidine blue and Giemsa methods. They are orthochromatic (a mixture of blue and red) with an Alcian blue–safranin method, 271 and orange-red using an enzymatic method (chloroacetate esterase). 272 In formalin-fixed material, toluidine blue may fail to stain up to 20% of mast cells.273. and 274. Carnoy’s medium appears to be a better fixative than formol saline for their demonstration. 271 Mast cells express tryptase, leukocyte common antigen (CD45), CD43, CD68, MCG-35, 275 3G5, 276 and CD117, the c-kit encoded tyrosine kinase receptor protein.277. and 278. Microphthalmia transcription factor (MITF) has also been detected in mast cells. 279
The role of genetics in mast cell proliferations has centered on the KIT proto-oncogene (formerly c-kit) which, as mentioned above, encodes a tyrosine kinase transmembrane receptor that is expressed on many cells including mast cells, hemopoietic stem cells, and also melanocytes. 280 Its role in piebaldism is mentioned elsewhere (see p. 282). The KIT mutations have been found in sporadic adult mastocytosis and in children at risk for extensive or persistent disease, but not in typical pediatric mastocytosis.281.282.283. and 284. The KIT gene has been localized to chromosome 4q11–q12.280. and 285.
In the skin, mast cells are usually found in the dermis with some accentuation around the superficial vascular plexus and appendages.274. and 286. They vary in number in the skin of different parts of the body, being most abundant in that of the scrotum. 287 They are more common in acral sites than in central sites such as the abdomen. 288 On average, there are approximately 7000 mast cells/mm3 of skin. 289 Mast cells in the mucosa are smaller and contain fewer granules than those in the dermis. 290 Mast cells are increased in the upper dermis in more than a third of healthy volunteers sitting in front of television and personal computer screens. 291 The significance of this finding is uncertain.
On ultrastructural examination the cytoplasm of mast cells contains 80–300 membrane-bound granules, which are modified lysosomes with a highly structured internal architecture and which appear in sections as whorls or scrolls. 292 Mast cell granules are larger in black skin than in white skin. This larger size appears to result from fusion of granules. 293 Other organelles include mitochondria, lipid droplets, microfilaments, and, rarely, melanosomes. 294 There are numerous microvilli projecting from the cell surface, and in mastocytosis these interdigitate with the projections of adjacent cells. 292
Mast cells, which are derived from a bone marrow stem cell that expresses CD34, produce a variety of pharmacologically active agents which may be preformed or which may form in the cytoplasmic granules in response to various stimuli.290.295.296. and 297. These agents include substances with vasoactive and smooth muscle contracting properties such as histamine, leukotrienes (B4, C4, D), and prostaglandin D2. These substances may be responsible for the pruritus, flushing, and syncope that sometimes occur in mast cell disease. 290 Mast cells also manufacture chemotactic factors, enzymes (neutral proteases, acid hydrolases), and heparin. Mast cell degranulation, with release of these substances, is a calcium-dependent process triggered by chemical, physical, and immunological stimuli. 295 Mast cells have numerous surface receptors for the Fc part of IgE, and this is responsible for mediating their immunological degranulation.295. and 298. One of these receptors (FcεRI), on stimulation, results in the release of an array of cytokines, including various interleukins, interferon-γ, tumor necrosis factor-α, and granulocyte–macrophage colony-stimulating factor. 299 Other categories of surface receptors are present, including cytokine receptors and integrins.
An aspect of mast cell physiology that has largely been ignored is that mast cells can secrete mediators without overt degranulation. 300 They are also involved in a variety of neuroinflammatory diseases, especially those worsened by stress. In these circumstances mast cells are activated by a variety of substances that stimulate mast cells to secrete mediators, without overt degranulation. 300
The exact role of mast cell growth factor (MGF) in humans remains to be clarified. It is produced by keratinocytes and fibroblasts. It stimulates not only mast cell proliferation but also melanocyte proliferation and melanin pigment production in vitro. 296 This might explain the hyperpigmentation overlying mast cell lesions.
Mast cells appear to participate in nearly all diseases of connective tissue. They are present in wound healing, keloids, chronic inflammation, parasitic infestations, urticarias, atopic eczema, lichen planus, psoriasis, Behçet’s syndrome, pretibial myxedema, scleroderma, and lichen simplex chronicus, to name just some of the conditions in which mast cells are increased.289.292. and 299. They are also increased in the stroma of neurofibromas and other neural tumors, and in mycosis fungoides and basal cell carcinomas. 292 Mast cells are sometimes found in the epidermis in various dermatoses. 301
Mast cell degranulation has been incriminated in the pathogenesis of the pruritus that occurs in polycythemia rubra vera. 302 This process has also been suggested as a possible cause of the pruritus in chronic renal failure, although in one study there was no correlation between mast cell numbers and the presence or absence of pruritus in patients undergoing hemodialysis for chronic renal failure. 303 Furthermore, antihistamines are unhelpful in controlling the symptoms.304. and 305.
Mastocytosis comprises a spectrum of related diseases in which there is an increase in mast cells in one or more organs. 290 It usually occurs as a sporadic disease that is often transient and limited in children and progressive in adults.283.306. and 307. There may be symptoms, such as pruritus, related to the release of various products from these cells.
Two KIT mutations at the 560 and 816 loci have been demonstrated to result in KIT autoactivation, and they are believed to be responsible for increased mast cells arising originally from the bone marrow.308. and 309. Patients with adult-onset mastocytosis and those with associated hematological diseases, usually express activating mutations of KIT. 310 Monoclonality has been reported. 311
The World Health Organization (WHO) variants of mastocytosis are shown in Table 40.3. This classification focuses on the hematological manifestations of the disease and not the cutaneous ones. 312 Accordingly, a more traditional classification will be used, as follows:
• Cutaneous mastocytosis
Urticaria pigmentosa
Solitary mastocytoma
Diffuse cutaneous mastocytosis
Telangiectasia macularis eruptiva perstans (TMEP)
• Systemic mastocytosis
With cutaneous lesions
With extracutaneous lesions only
• Malignant mast cell disease
Malignant mastocytosis
Mast cell leukemia.
Urticaria pigmentosa (maculopapular cutaneous mastocytosis in the WHO classification) is the most common clinical variant of mastocytosis, accounting for approximately 80% of all cases.310.313. and 314. It usually presents as a generalized eruption of multiple red-brown macules, or rarely papules, predominantly affecting the trunk, but sometimes also the extremities and head. There may be few lesions, which are widely scattered, or hundreds. They are pruritic in less than 50% of cases, although in the majority of cases lesions will develop a wheal and flare when rubbed (Darier’s sign). 287 Onset of urticaria pigmentosa is in the first 4 years of life in 75% of cases. 315 Childhood cases have a good prognosis, usually without the occurrence of systemic involvement.316. and 317. Most lesions clear by puberty in 80% of affected individuals, although lower clearance rates have been reported. 318 Vesiculation is a common transient change in lesions of infancy and childhood.319. and 320. Adult-onset disease is marked by persistence of lesions, and the development of systemic disease in approximately 40% of cases.295.321.322. and 323. Adults with extensive cutaneous disease experience more pruritus and flushing than those with less extensive disease. 324 Bone marrow involvement is common in adults with cutaneous mastocytosis. 325 A scoring system that reflects the extent of cutaneous involvement, and the symptoms and activity of lesions has been developed. 326
Rare clinical presentations of urticaria pigmentosa include a congenital or early-onset bullous form,282.327.328.329.330.331. and 332. unilateral swelling of the vulva, 333 a nodular variant, 309 a scarring alopecia, 334 the presence of yellowish lesions resembling xanthoma or pseudoxanthoma elasticum (xanthelasmoid mastocytosis),335.336.337.338. and 339. and urticaria, dermatographism, 340 massive peripheral eosinophilia, 341 or intractable pruritus without any obvious lesions. 342 Bullous mastocytosis resembling staphylococcal scalded skin syndrome has been reported. 343 Urticaria pigmentosa has been associated with multiple myeloma, 344 germ cell tumors of the ovary, 345 Wilms’ tumor, 346 HIV infection, 347 bilateral neurosensory hearing loss, 348 and juvenile xanthogranuloma. 349 Sporadic familial cases occur.328.350.351.352. and 353.
The primary objective of treatment in all categories of mastocytosis is to control symptoms of mast cell ‘degranulation’ and to block mast cell proliferation. 354 Both these goals are achieved with PUVA therapy, and with UVA-1 phototherapy. 354 In systemic mastocytosis, therapy with the tyrosine kinase inhibitor imatinib inhibits KIT associated with the FIP1L1–PDGFRA fusion oncogene but not the more common KIT D816V mutation.309. and 355. Other treatment options for systemic disease are beyond the scope of this book; they have been reviewed elsewhere.355. and 356.
Solitary lesions account for approximately 10% or more of childhood mastocytoses.306. and 357. They may occur anywhere on the body, but there is a predilection for the trunk and wrists. The palm is a rare site of involvement.358. and 359. Small solitary lesions are sometimes called ‘mast cell nevi’, and the term ‘mastocytoma’ is reserved for larger nodular lesions, which may measure up to 3 cm in diameter. 295 A combined mastocytoma–junctional nevus has been reported. 360 It may have resulted from the synchronous proliferation of two cell types in the one field rather than being a collision tumor. 360 Solitary mastocytomas may be associated with extralesional symptoms including pruritus, flushing, headaches, and gastrointestinal symptoms. 361 Solitary lesions tend to involute spontaneously, and there is usually no indication for surgical removal. This regression is mediated by loss of proliferating activity of mast cells, an increase in apoptotic mast cells, and increased expression of stem cell factor in remaining mast cells. 362 The latter finding is difficult to explain.
Diffuse cutaneous mastocytosis is a rare variant that usually begins in early infancy with thickening of the skin, which may be erythematous or yellow-brown in color.310.363.364.365. and 366. Pruritus and blistering are common, and the bullae that form are more persistent than in urticaria pigmentosa of childhood. Leathery (pachydermatous) lesions may be present.283. and 367. Nodules may develop within the thickened skin, but this does not necessarily indicate a bad prognosis. 368 Systemic involvement is common. 283
TMEP is the universally accepted short designation for telangiectasia macularis eruptiva perstans, a rare adult form of mastocytosis with a high incidence of systemic involvement.369. and 370. Childhood cases and two familial series with childhood onset have been reported.371.372.373. and 374. Erythema and telangiectasia are found in faintly pigmented macules on the trunk and proximal parts of the extremities. 270 Facial involvement has been reported.375. and 376. Multiple myeloma is a rare association. It has been suggested that aberrations in the KIT pathway may explain the abnormal proliferation of both lineages. 377 TMEP has also been associated with a myeloproliferative disorder. 378
In systemic mastocytosis there is a proliferation of mast cells in various tissues apart from, or in addition to the skin. 379 Systemic mastocytosis may develop in childhood cases of urticaria pigmentosa that persist beyond puberty, and in approximately 40% of adults with urticaria pigmentosa, usually of long standing.315.321. and 380. It may also be associated with TMEP.381. and 382. A case of systemic mastocytosis with diffuse cutaneous involvement and hematological disease presenting in utero has been reported. 383 Cutaneous lesions are most common on the trunk, although all skin areas, including mucous membranes, may be involved. Papillomatous and verrucous lesions are rare findings. 384 Recurrent syncope and anaphylaxis were the presenting features of one pediatric patient. 385 Thrombocytosis is another rare manifestation of systemic mastocytosis. 386
The bone marrow is the tissue most frequently involved in systemic mastocytosis, followed by the liver, spleen, gastrointestinal tract, lymph nodes and, rarely, other organs.379. and 387. Sometimes bone marrow is the only tissue involved besides the skin. Systemic mast cell disease without skin involvement is quite uncommon, and often difficult to diagnose.369. and 379. However, the urinary excretion of the histamine metabolite methylimidazoleacetic acid is increased in cases of systemic mastocytosis.290. and 388. Flow cytometry on bone marrow samples, and the use of serum or marrow-blood tryptase levels may assist in making a diagnosis in indolent cases without cutaneous involvement.324.389. and 390.
Systemic mastocytosis may progress to malignant mastocytosis and/or mast cell leukemia. Various lymphoproliferative and myeloproliferative conditions may sometimes eventuate,391.392. and 393. particularly myelogenous leukemia.369.394. and 395. It has also been associated with a lymphocytic lymphoma. 396 The hypereosinophilic syndrome is a rare association. 397 Cutaneous lesions regress in approximately 10% of older patients who have systemic mastocytosis. 398 In patients with an associated hematological condition, this regression may be accompanied by progression of the hematological disease. 398 In patients with systemic mast cell disease without associated hematological disorders, the bone marrow mast cell burden, bone marrow eosinophilia, and serum alkaline phosphatase levels are of prognostic significance. 399 It has been claimed that up to one-third of individuals with systemic mastocytosis may progress to malignancy, but this seems unduly pessimistic in the light of other studies, one of which showed that the clinical course of systemic mastocytosis was stable over a period of 10 years in all those followed. 321
A small proportion of cases are clonal. This is more common in cases with associated eosinophilia. They may carry the FIP1L1–PDGFRA fusion oncogene, which results from an interstitial deletion of chromosome 4q12, thereby generating an active PDGFRA tyrosine kinase. 355
There may be a progressive proliferation of atypical mast cells leading to the enlargement of various organs. This usually follows systemic mastocytosis, and has been called malignant mastocytosis and mast cell reticulosis. 400 The extremely rare mast cell sarcoma is a localized mass of malignant mast cells in the soft tissues.400. and 401. Mast cell leukemia may develop in any of these settings or as a progression of systemic mastocytosis. 400 There is extensive bone marrow infiltration and atypical mast cells circulating in the peripheral blood. 402 It has a poor prognosis. 270
The histological pattern of mastocytosis is similar regardless of the clinical type, although there are major variations in the number of mast cells present. 402 The infiltrate is predominantly in the upper third of the dermis, at times in proximity to the dermoepidermal junction (Fig. 40.5). 402 In the usual macular lesions of urticaria pigmentosa the infiltrate may vary from sparse and perivascular to larger aggregates of mast cells. Perivascular mast cells may be cuboidal or fusiform in shape, whereas those in larger aggregates tend to be cuboidal (Fig. 40.6). A scattering of eosinophils is usually present, and there may be superficial edema in lesions that are rubbed prior to removal. Basal hyperpigmentation is a useful clue to the diagnosis of urticaria pigmentosa and some other types of mastocytosis.
Urticaria pigmentosa. (A) Mast cells fill the papillary dermis. (B) A higher power view of the mast cells. (H & E)
Urticaria pigmentosa. Numerous mast cells are present in the upper dermis. There is also mild hyperpigmentation of the basal layer. (Toluidine blue)
In solitary mastocytoma there are dense aggregates of mast cells in the dermis, sometimes extending into its deeper levels and even into the subcutis (Fig. 40.7). 270 Cells with bilobed or multilobed nuclei were noted in one case. 403 Eosinophils may be present in small numbers; massive infiltration is a rare occurrence.404. and 405. Localized necrobiosis and stromal fibrosis have been reported.406. and 407. In TMEP there may be only subtle alterations in mast cell numbers; the cells tend to be fusiform and loosely arranged around the dilated vessels of the superficial plexus (Fig. 40.8). Eosinophils are usually absent. In diffuse cutaneous mastocytosis there are loosely arranged mast cells throughout the dermis. 368 Fibrosis is sometimes present. 367 The mast cells are deeper in the dermis in the xanthomatous form.
Solitary mastocytoma. (A) Mast cells fill the dermis. (H & E) (B) The cells express CD117. (Immunoperoxidase stain)
Telangiectasia macularis eruptiva perstans (TMEP). (A) The mast cells are predominantly perivascular in location. The blood vessels are not as telangiectatic as usual. (H & E) (B) Mast cells surround dilated vessels in the upper dermis. (Immunoperoxidase stain for CD117)
Superficial edema leading to subepidermal vesiculobullous changes is common with mast cell lesions of infancy and childhood. 408 There may be eosinophils, mast cells, and occasional neutrophils within the bullae, and a diffuse aggregate of mast cells in the upper dermis below the band of edema or the blister cavity.
Quantitative studies have shown that the number of mast cells in the cutaneous lesions of mastocytosis is from 2 to 160 times that in the adjacent normal skin.273.409.410. and 411. Normal skin may contain up to 20 mast cells per high-power (×40) field. In TMEP, in which the increase may be subtle, it is often useful to have some normal skin at one end of the biopsy for comparison with lesional skin. 270 Qualitatively, the mast cells in mastocytosis resemble normal mast cells, with little atypia and only minor changes detected by morphometry.412. and 413. They give the staining reactions described for normal mast cells; the choice of stain (toluidine blue, astra blue, Giemsa or chloroacetate esterase) often depends on individual preference. Mast cell tryptase can also be used.307. and 414. Immunohistochemistry for CD117 and CD68 has been used.277. and 415. CD117 gives a clearer staining pattern than some of the earlier histochemical methods. Microphthalmia transcription factor (MITF) is not commonly sought. 416 Nevertheless, its presence, along with staining for mast cell tryptase, effectively discriminates mast cell disease from myeloid leukemia cutis. 417 In some cases of malignant mastocytosis the cells lack metachromatic granules and fail to stain with routine methods. The antitryptase antibody G3 will stain mast cells in these circumstances. 418
The expression of CD25 on cutaneous mast cells from adult patients presenting with urticaria pigmentosa is predictive of systemic mastocytosis. 419
The ultrastructural features of normal mast cells have been described earlier. In mastocytosis the cells have prominent surface projections which interdigitate with adjacent cells. Other reported findings include giant cytoplasmic granules,420. and 421. and mast cells with endocytic and autophagic vacuoles. 422 A blue nevus combined with a mastocytoma has been reported, with some cells containing both melanosomes and mast cell granules. 423
Brachioradial pruritus is a rare dermatosis that presents with a chronic intermittent intense pruritus localized to the elbow region in the vicinity of the brachioradialis muscle.424.425.426. and 427. Sometimes the lateral surface of the upper part of the arm, just below the midpoint, is involved.428. and 429. The pruritus, which may be unilateral or bilateral, is often accompanied by a burning sensation. 424 The itch feels ‘deep’. It progressively worsens towards evening. 430 The application of ice packs provides relief. This is a diagnostic feature, not a recommended treatment. 431 Several members of the one family have been affected. 432 Their disease was characterized by symptom-free periods. Affected individuals are sometimes referred to a psychiatrist because there is little to see clinically except for mild poikilodermatous mottling suggestive of solar damage. 429
This condition is seen in white people living in tropical and subtropical climates, suggesting that exposure to the sun is of some etiological importance.424. and 433. It has therefore been regarded in the past as a solar dermopathy and part of the spectrum of solar pruritus.434.435.436. and 437. However, in some patients clinical improvement has been achieved by cervical spine manipulation, suggesting that in these patients there may be a component of nerve damage from cervical spine disease.438.439. and 440. It has also been associated with a spinal cord tumor. 441 The current consensus view is that ultraviolet radiation may act as a trigger in some patients, although underlying neurological cervical injury is also an important etiological factor.442.443. and 444. In one series, the disease could be attributed to a neuropathy in all patients. 445 Its etiological similarity to notalgia paresthetica (see p. 298) has been noted. 446
Neuropathic scrotal pruritus is another similar condition. It is due to a lumbosacral radiculopathy. 447
Brachioradial pruritus is often refractory to treatment with topical or oral corticosteroid, and antihistamines. A controlled trial showed no benefit from capsaicin. 448 Several cases have been successfully treated with gabapentin, an anticonvulsant used also in the treatment of neuropathic pain. 448
Little may be observed on a casual examination of a biopsy. Closer inspection will usually reveal mild, patchy hyperpigmentation of the basal layer with minimal melanin incontinence, mild telangiectasia of superficial blood vessels, and occasional mononuclear cells around dermal vessels and appendages (Fig. 40.9). 429 There is also some solar elastosis.
Brachioradial pruritus. A few lymphocytes and mast cells surround the telangiectatic vessels in the upper dermis. (H & E)
Stains for mast cells reveal that many of the mononuclear cells are mast cells, and although their increase in number is marginal, they are usually quite plump and contain numerous cytoplasmic granules. 429 This applies particularly to the mast cells in the vicinity of the pilosebaceous follicles and eccrine glands. This is one reason for its inclusion here.
The histiocytic infiltrates are a heterogeneous group of disorders which were also known, in the past, as the ‘non-X histiocytoses’ because the infiltrating cells lack Birbeck granules (Langerhans bodies) and other markers of Langerhans cells, which are the cells found in histiocytosis X.449.450. and 451. Contemporary classifications now regard Langerhans cells as one class of histiocyte, indicating that our concepts have gone ‘full circle’. For convenience, the Langerhans cell histiocytes will be considered later in this chapter.
The histiocyte is a somewhat controversial cell which still lacks precise definition.452.453.454. and 455. The term ‘histiocyte’ has been used in the past for almost any cell with a reniform or indented nucleus, a diameter of 10–25 µm, and a nuclear/cytoplasmic ratio of about 1:1. 452 The advent of immunohistochemistry, with the use of various monoclonal antibodies, has allowed the more precise characterization of cells that resemble each other morphologically. These studies have confirmed that the term ‘histiocyte’ has been used in the past for a variety of cells, including Langerhans cells and other immune accessory cells and certain subsets of T and B lymphocytes. 452
In 1997, the Histiocyte Society in conjunction with the WHO Committee on Histiocytic/Reticulum Cell Proliferations published what they called a ‘Contemporary Classification of Histiocytic Disorders’. 456 They defined histiocytes as ‘a group of immune cells, familiar to morphologists, that includes macrophages and dendritic cells’. They pointed out that macrophages and dendritic cells were polar representatives of one common regulatory system. The origin of the various histiocytes is still controversial but it appears that the macrophage, the indeterminate cell and the Langerhans cell are derived from the CD34+ hemopoietic progenitor, while the dermal dendrocyte is derived from cutaneous mesenchymal (fibroblastic) precursors, although a hemopoietic source has also been proposed. 456 The various types of histiocyte express different antigens that assist in their distinction. They are shown in Table 40.4.
CD45, CD14, CD68, CD163, lysozyme positive
Also, often CD11b, CD11c, HAM-56, Mac387 positive S100, CD1a, factor XIIIa negative
Factor XIIIa, CD45, CD68 positive
S100, CD1a negative
CD45, S100, CD1a positive
Factor XIIIa negative (?)
Birbeck granules absent
CD45, S100, CD1a, CD101 positive
Factor XIIIa negative
Birbeck granules present
Common usage today considers the histiocytes as tissue macrophages derived from blood monocytes. They act as professional phagocytes or antigen-presenting cells. They can have different phenotypes depending upon the tissue in which they are found. 457 Both phagocytic and antigen-presenting histiocytes are able to produce inflammatory cytokines and mediators. 457
Whereas Langerhans cells and indeterminate cells stain positively for S100 protein and CD1a, cells of the monocyte–macrophage system stain with the CD11B, CD11C, CD14, CD68, HAM-56, and Mac387 antibodies, although Mac387 lacks sensitivity.456.458.459. and 460. Lysozyme may also be present in these cells. Another marker, MS-1, a high-molecular-weight extracellular protein specific for sinusoidal endothelial cells and dendritic perivascular macrophages, is expressed by the cells of the various histiocytic tumors but not by Langerhans cells or the palisading histiocytes of granuloma annulare. 461 Although the non-Langerhans histiocytes are usually considered to be S100 negative, scattered cells in some tumors may be positive. 462
As further immunohistochemical studies come to hand, using expanded panels of monoclonal markers, it may be necessary to recategorize some of these entities. 463 Furthermore, some cases have been reported which defy orderly classification, despite being studied by means of various immunohistochemical markers.464.465.466.467.468. and 469.
As mentioned above, a new classification of histiocytic disorders was published by the Histiocyte Society/WHO group in 1997. 456 As it applies to all body systems, a modified version covering the skin is shown in Table 40.5. It can be seen that many of the diseases to be discussed in this section are dismissed as ‘related disorders’ of juvenile xanthogranuloma. Unfortunately, it is difficult to classify all of these conditions into one of the various categories of the histiocytoses because of conflicting immunohistochemical results for some of the rarer entities. An attempt will be made to do this below, based on current knowledge. Before doing so, it is worth noting the approach of Zelger and colleagues to this confusing topic. 470 They have attempted a morphological classification of most of the non-Langerhans histiocytoses of presumed dendritic origin. A modified version of their classification is shown in Table 40.6.
Juvenile xanthogranuloma (mononuclear type)
Benign cephalic histiocytosis
Generalized eruptive histiocytosis
Progressive mucinous histiocytosis
Papular xanthoma
Xanthoma disseminatum (rare)
Spindle-cell xanthogranuloma
Progressive nodular histiocytosis
Progressive mucinous histiocytosis
Juvenile and adult xanthogranuloma
Reticulohistiocytoma
Progressive mucinous histiocytosis
The various non-Langerhans histiocytoses will be discussed in the order given below. The categorization is based on the author’s interpretation of the current Histiocyte Society/WHO classification and supplemented by published papers,456.470. and 471. notwithstanding the problems posed by some apparent discrepancies in published immunohistochemical results. It should be noted that a comparatively recent paper has suggested that the plasmacytoid monocyte, rather than the dermal dendrocyte, is the cell of origin of juvenile xanthogranuloma. 472 Other tumors in this same category would presumably have a similar origin.
• Non-Langerhans histiocytoses of dendritic origin
Juvenile xanthogranuloma (including spindle-cell xanthogranuloma)
Benign cephalic histiocytosis
Progressive nodular histiocytosis
Xanthoma disseminatum
Erdheim–Chester disease
Sea-blue histiocyte syndrome (?)
Generalized eruptive histiocytoma
Progressive mucinous histiocytosis
Multicentric reticulohistiocytosis
Reticulohistiocytoma
Familial histiocytic dermatoarthritis
• Non-Langerhans histiocytoses of disputed/uncertain origin
Necrobiotic xanthogranuloma
Cutaneous atypical histiocytosis
Indeterminate cell histiocytosis
• Histiocytoses of macrophage origin
Rosai–Dorfman disease
Giant-cell rich histiocytic dermatitis/panniculitis
Hemophagocytic lymphohistiocytosis
• Malignant histiocytoses
Histiocytic sarcoma
• Miscellaneous
Reactive histiocytoses
Intravascular/intralymphatic histiocytosis.
Regressing atypical histiocytosis was formerly included with this group, but is now regarded as a T-cell lymphoma on the basis of immunohistochemistry.
It is becoming increasingly apparent that there is considerable overlap between the clinical and histopathological features of the various cutaneous histiocytoses, suggesting that they possibly represent one disease ‘entity’ with a wide spectrum of clinical presentations, rather than many discrete disorders.473.474. and 475. This is particularly so for the non-Langerhans histiocytoses of presumed dendritic origin. Three clinical groups of non-Langerhans cell histiocytoses have been proposed (see Table 40.7). 476
Erdheim–Chester disease
Rosai–Dorfman disease
Sea-blue histiocyte syndrome
Cases reported variously as reactive angioendotheliomatosis, intravascular histiocytosis, and cutaneous histiocytic lymphangitis consist of dilated vascular channels containing histiocytic cells. They are considered together as intravascular/intralymphatic histiocytosis at the conclusion of the histiocytic infiltrates (see p. 961).
Juvenile xanthogranuloma is a normolipemic histiocytosis composed of cells originally thought to be derived from dermal dendrocytes.449.456.477.478. and 479. However, a subsequent study has suggested that the plasmacytoid monocyte is a more likely precursor. 472 Solitary or multiple red-brown papulonodules, 1–10 mm in diameter or larger,480.481. and 482. are found on the head and neck, upper part of the trunk, and proximal parts of the limbs.483. and 484. In one series, a solitary cutaneous lesion accounted for 67% of cases; multiple cutaneous lesions were present in 7% of the patients. 485 In the series from Kiel, 81% were solitary. 486 The sole of the foot, 487 vulva, 488 anogenital region, 489 finger, 490 and proximal nail fold491 are rare sites. Atypical forms with extensive facial, 492 or generalized eruptions493.494. and 495. have been documented. A thickly crusted variant on the scalp has been described. 496 Plaque-like and clustered lesions also occur.497.498. and 499. A linear distribution is another pattern.500. and 501. In two-thirds of all cases onset is within the first 6–9 months of life;502.503.504. and 505. approximately 5–35% are present at birth.486.506. and 507. Severe congenital systemic juvenile xanthogranuloma has been reported in monozygotic twins. 508 Late onset in adolescence or adult life occurs in approximately 10–30% of cases.509.510.511.512.513. and 514. The term ‘xanthogranuloma’ or ‘adult xanthogranuloma’ is used for these cases. There is a male predominance. 485 Spontaneous involution usually occurs after many months or even years; lesions persist in some individuals who develop their first lesions after the age of 20 years.515. and 516.
A small percentage of patients have ocular involvement;502. and 517. visceral, including oral, involvement is exceedingly rare.477.493.518.519. and 520. Rare cases involving a peripheral nerve have been reported. 521 One case arose in an organoid nevus (nevus sebaceus), 522 and another in the substance of the breast. 523 Juvenile xanthogranuloma has been reported in association with neurofibromatosis (sometimes with associated juvenile chronic myelogenous leukemia),524.525.526.527.528. and 529. other hematological malignancies,530. and 531. Niemann–Pick disease (see p. 486), 532 contralateral lymphadenopathy with a histiocytic infiltrate, 533 adult T-cell leukemia/lymphoma, 534 and urticaria pigmentosa. 535 Juvenile xanthogranuloma has developed in three children as a sequel to Langerhans cell histiocytosis. 536 In another case an eruptive variant developed following treatment of Langerhans cell histiocytosis with chemotherapy. 537 A case resembling juvenile xanthogranuloma developed in a boy at puberty who had homozygous familial hypercholesterolemia, under treatment with low-density lipoprotein apheresis and drug therapy. 538
Although the etiology of juvenile xanthogranuloma is unknown, studies have shown that cholesterol is the principal lipid in the lesions. 539 Furthermore, there are no unusual sterols present. 539 A recent study demonstrated clonality in one case. 540 This remains to be confirmed.
Simple tumor excision is the treatment of choice except in the very rare systemic form, in which multimodal chemotherapy is indicated. 486
There is a nodular, poorly demarcated dense infiltrate of small histiocytes involving the dermis and sometimes the upper subcutis as well. Rarely, deep extension into skeletal muscle is present.542. and 543. The cells are polygonal or spindle-shaped and plump, and have indistinct cytoplasmic borders. Mitoses are rare. Whereas early lesions are fairly monomorphous, with inconspicuous foam cells,544.545.546.547.548. and 549. mature lesions contain foamy histiocytes and varying numbers of Touton cells (Fig. 40.10 and Fig. 40.11). These cells have a less foamy periphery than those seen in other xanthomatous conditions. They are transient elements, not present in every case, particularly in extracutaneous sites. 550 Small aggregates of foam cells may be seen in some cases, and fully xanthomatized variants also occur (Fig. 40.12). There are also scattered lymphocytes and neutrophils, rare plasma cells, and sometimes eosinophils. Lesions of longer duration will show interstitial fibrosis and proliferating fibroblasts.
Juvenile xanthogranuloma. There is an admixture of several cell types forming a dense infiltrate. (H & E)
Juvenile xanthogranuloma. The Touton giant cells, which are unusually large, are admixed with lymphocytes and histiocytes. (H & E)
Juvenile xanthogranuloma. There is prominent xanthomatization with epidermal atropy. (H & E)
Based on the review of 129 cases in the Kiel Pediatric Tumor Registry, 486 it appears that there is a time-dependent course in the evolution of lesions. There is an early, predominantly mononuclear infiltrate, followed by the classic Touton cell-rich stage. Finally there is a stage with a spindle-cell pattern with a variable component of mononuclear and multinucleated cells.486. and 550.
The lesion reported as a spindle-cell xanthogranuloma is best regarded as a histological variant of the adult form of xanthogranuloma. It is composed of spindle-shaped histiocytes arranged in a storiform pattern. Other mononuclear (xanthomatized, oncocytic, vacuolated, and scalloped) and multinucleate (Touton) histiocytes are present. 551
The scalloped-cell xanthogranuloma is another variant in which there is a predominance (>75% of all cells) of scalloped histiocytes. Such cells have an angulated or scalloped border, set apart from one another in a delicate fibrous matrix. 552 The cytoplasm of the cells is homogeneous and slightly eosinophilic while the nucleus is round to oval and large.
Fat stains confirm the presence of some lipid. Hemosiderin has been demonstrated in a few cases. 553 The histiocytes are usually positive for CD68, HAM-56, HHF35, cathepsin B, vimentin, factor XIIIa, fascin, CD4, lysozyme, and α1-antichymotrypsin, 483 but negative for Mac387, smooth muscle actin, CD34, CD1a, and S100 protein.493. and 541. Approximately 1–10% of the other cells in any lesion are S100 positive. 554 Such cells are elongated or dendritic in appearance. 509 Staining for the various markers listed above may be more intense at the periphery of the lesion. Membranous staining is the predominant pattern seen with CD10. 555
It has been suggested that the deep forms are derived from dermal indeterminate cells (CD1a positive, S100-protein negative), but this is based on only one study. 556 These two markers were not studied in a recent series of three deep cases; the cells were CD68 and factor XIIIa positive. 557
Ultrastructural examination of mature lesions has shown lipid vacuoles (often without a limiting membrane), lysosomes, cholesterol clefts and myeloid bodies, but no Birbeck granules. Early lesions have only a few lipid-laden cells and there are complex interdigitations of the cell membrane. 560 Fibroblastic cells have also been noted in some lesions. 561
Benign cephalic histiocytosis is a clinically distinct non-lipid ‘non-X’ histiocytic proliferation in children.562.563.564. and 565. Fewer than 50 cases have been reported. 566 It is characterized by the development of asymptomatic red-brown maculopapules, 2–5 mm or more in diameter, on the face, whence the condition may evolve to affect the neck, upper part of the trunk, arms, and other parts of the body.567.568. and 569. The palmoplantar regions, mucous membranes, and viscera are spared; 562 however, a case with diabetes insipidus has been reported. 570 In another case, insulin-dependent diabetes mellitus developed. 571 Spontaneous regression, complete or partial, occurs during childhood, without scarring.562. and 572. Similar lesions have been reported in an adult with T-cell lymphoma. The exact status of this case is uncertain. 573 There is increasing evidence that benign cephalic histiocytosis is a clinicopathological variant of juvenile xanthogranuloma,574. and 575. or part of a spectrum with it. 576
There is a diffuse infiltrate of histiocytes, mainly in the upper dermis and in close apposition to the undersurface of the epidermis. The cells have an oval or reniform vesicular nucleus and ill-defined pale cytoplasm. 563 There is usually no cytoplasmic lipid in the early stages of evolution, but xanthomatization can be seen in lesions of long duration. 574 Occasional multinucleate histiocytes are present. There are scattered or grouped lymphocytes within the infiltrate, and occasional eosinophils in some lesions. The histiocytes are negative for S100 protein and CD1a, but positive for CD68, CD11b, CD14, HAM-56, and factor XIIIa.562.572. and 574.
The most characteristic feature of the histiocytes is the presence of many coated vesicles, 500–1500 nm in diameter, in the cytoplasm. Comma-shaped inclusion bodies are present in 5–30% of the histiocytes. 577 No Birbeck granules or lipid droplets are present. Desmosome-like junctions are present between the histiocytes in densely cellular areas. 562
Progressive nodular histiocytosis is a normolipemic non-Langerhans cell histiocytosis in which multiple yellowish-brown papules and nodules develop on the skin and mucous membranes.578.579.580. and 581. The nodules are more common on the trunk, while the papules are widely and randomly distributed on the body. Both types of lesion can occur around the genitalia. 582 Similar lesions have been noted in a patient with acute myelomonocytic leukemia, 583 and in another with chronic myeloid leukemia. 584 It has also been reported in a patient with a hypothalamic tumor. 585 It has been suggested that all these cases are variants of, or closely related to, (juvenile) xanthogranuloma. Another confusing feature is the use of the term ‘progressive nodular histiocytosis’ for cases with morphological features of multicentric reticulohistiocytosis but an absence of the usual systemic symptoms of this disorder (see below). 478 Leonine facies have been present in the latter condition.478.586. and 587. In the initial reports of the entity under discussion, the word ‘histiocytoma’ was used instead of ‘histiocytosis’ in the title.
The nodules have a similar appearance to the cellular and fibrous patterns seen in a dermatofibroma. There are histiocytes, foam cells, and spindle-shaped cells, sometimes arranged in a storiform pattern. 551 The cells are embedded in a delicate fibrocollagenous matrix. Touton cells may be present. There are usually no other inflammatory cells. Fat and hemosiderin can be demonstrated in the histiocytes with special stains.
Xanthoma disseminatum is a rare normolipemic histiocytic proliferation of dendrocyte origin. It presents with papules, nodules, and plaques that have a predilection for flexural areas, the proximal parts of the extremities, and the trunk. The face is sometimes involved. 588 The lesions are initially reddish-brown in color but become yellow with time. 478 Mucosal lesions and transitory diabetes insipidus are present in approximately 40% of affected individuals.589.590.591. and 592. Skeletal involvement has also been recorded. 593 Waldenström’s macroglobulinemia was present in one case. 594 The condition runs a chronic but usually benign course. Widespread systemic involvement and death have been reported.595. and 596.
Xanthoma disseminatum appears to be a clinically distinctive disorder in which there is a primary proliferation of histiocytes with subsequent accumulation of lipid.597. and 598. It may represent an evolutionary form of generalized eruptive histiocytoma (see below) and as such be part of the spectrum of dendritic disorders related to (juvenile) xanthogranuloma.
Treatment with CO2 laser therapy has been successful. 599 Azathioprine and corticosteroids were used in a child with xanthoma disseminatum who had hepatic involvement. 600 In another patient, a combination of three lipid-lowering agents was used. 601
Histiocytes predominate in the early stages, but in established lesions there are histiocytes, foam cells, spindle cells, Touton cells, and a moderate number of chronic inflammatory cells. 478 There may be phagocytosis of elastic fibers and sometimes of collagen by macrophages. 597 Siderosis is often observed, and this is quite prominent in the rare variant known as xanthosiderohistiocytosis. 602
Immunohistochemical characterization of the cells has shown HLA-DR positivity but no staining for S100 protein, CD4 or CD1a. 603 The cells express CD68, Ki-M1p, CD14, CD11b, CD11c, and factor XIIIa.601.604. and 605. There is variable staining with Mac387.
The cells are similar to those seen in papular xanthoma and juvenile xanthogranuloma, but the plasma membranes of the foamy cells show many microvilli. 478 There are no Birbeck granules.
Erdheim–Chester disease is a rare, disseminated form of non-Langerhans cell histiocytosis. The age of presentation is typically in the fifth decade of life. 606 It is primarily a disease of long bones producing patchy medullary sclerosis on radiographs, with sparing of the epiphyses.582. and 607. Extraskeletal manifestations can occur in almost every organ including the lungs, pericardium, hypothalamus/pituitary, the orbit, and retroperitoneum.
The skin is affected in about 30% of cases. 606 Cutaneous lesions include diffuse dermal nodules similar to xanthoma disseminatum, 608 xanthelasmas, subcutaneous nodules, intertrigo-like lesions, 606 pretibial dermopathy, and pigmented patches on the lips and oral mucosa. Verruca plana-like papules, infiltrated by foamy cells, may also be present. 607
Erdheim–Chester disease is basically a xanthomatous process, but it has been considered here, perhaps arbitrarily, because it is primarily an infiltrate of histiocytes of non-Langerhans cell type. The variably lipidized histiocytes involve the dermis, often with extension into the subcutis. Sometimes there are Touton giant cells, but multinucleate cells may be completely absent. There is variable fibrosis; sometimes lesions are quite sclerotic. The cells are CD68+, factor XIIIa+, CD1a−, and S100−.
The sea-blue histiocyte syndrome (OMIM 269600) is a rare, inherited systemic histiocytosis characterized by histiocytes with deep azure blue granules in the cytoplasm on Giemsa, toluidine blue, and May–Gruenwald staining.457. and 582. Secondary forms, associated with storage diseases such as Niemann–Pick disease (type B), and other diseases have now been reported. 457 Commonly involved organs are bone marrow, liver, and spleen. Cutaneous lesions are rare and found in only a few patients. 582 They consist of nodular lesions, and brown pigmented macules, both on the face. When disseminated lesions are present, it may lead to death.
A partial deficiency of sphingomyelinase is a possible cause. 582 Two reports have linked the disease to the APOE gene at 19q13.2, which is involved in lipid metabolism (see OMIM log).
Cutaneous lesions consist of dermal infiltrates of large monomorphous histiocytes with cytoplasmic vacuoles and granules.457. and 582. The granules are yellow-brown on H & E, and dark blue with toluidine blue or Giemsa staining. They show yellow autofluorescence, and display birefringence under polarized light.
The histiocytes are CD68+ and S100−. They do not appear to have been tested for CD1a or factor XIIIa. 582
Generalized eruptive histiocytoma is a rare histiocytosis, characterized clinically by the development of recurrent crops of hundreds of small reddish papules which are distributed symmetrically on the trunk and on the extensor surfaces of the extremities.609.610.611.612.613. and 614. Approximately 30 cases have been reported. It has been documented in children as well as in adults.609.610.615. and 616. Lesions usually subside spontaneously, leaving macular hyperpigmentation or no residual features. It has been suggested that regression involves apoptosis of the infiltrating histiocytes. 617 In one case resolution followed exanthem subitum (roseola infantum). 618 Two cases have been associated with acute leukemia. 619 Evolution into xanthoma disseminatum has been reported, further evidence of the close interrelationship of the various non-Langerhans cell histiocytoses, which appear to form part of a continuous spectrum.620. and 621.
Although often thought to be a self-limiting disease, recurring crops of lesions continued for 12 years in one patient. This necessitated treatment, which was successful using a combination of corticosteroids, hydroxychloroquine and thalidomide. 622
There is an infiltrate of histiocyte-like cells, often spindle shaped, in the upper and mid dermis. Sometimes the cells are arranged in nests around blood vessels. They have pale cytoplasm and an oval nucleus. No lipid, iron, or PAS-positive material is demonstrable in the cytoplasm. A few lymphocytes, and sometimes fibroblasts, are intermingled but there are usually no giant cells or xanthoma cells, 620 but giant cells have been reported. 621 The histiocytes are negative for S100 protein and CD1a, but are positive for CD11b, CD14, CD36, factor XIIIa, CD68, and MS-1 protein.610.620.623. and 624. This latter antigen has recently been found in all of the non-Langerhans cell histiocytoses tested. It is specific for sinusoidal endothelial cells and dendritic perivascular macrophages. 461 The cells in generalized eruptive histiocytoma show only patchy positivity for Mac387. 623 A case with some S100-positive cells has been regarded as an indeterminate cell variant. 625
Benign cephalic histiocytosis and a case reported as ‘benign non-X histiocytosis’626 have features on light microscopy that are similar to those of generalized eruptive histiocytoma, but they differ clinically and ultrastructurally. 610 As mentioned earlier, many of the non-Langerhans histiocytoses of dendritic origin may be different expressions of the same histiocytosis. 611
Progressive mucinous histiocytosis (OMIM 142630), a very rare, non-Langerhans cell histiocytosis of childhood, was first described by Bork and Hoede in 1988.627. and 628. Sporadic cases in adults have since been reported. 629 Fewer than 20 patients have been described. It is characterized by a progressive eruption of skin-colored to red-brown papules with a predilection for the face and extremities. It appears to have an autosomal dominant mode of inheritance. 630 The gene responsible has not been elucidated.
There is a dermal infiltrate of epithelioid and spindle-shaped histiocytes, with the latter predominating in older lesions. 631 There is marked metachromasia of these cells with toluidine blue. 582 There are rare giant cells but no foam cells. 632 The cells are set in a collagenous stroma containing abundant mucin (Fig. 40.13). Mast cells can be quite numerous. 633 The cells stain for factor XIIIa, CD68, HAM-56, lysozyme, and vimentin, but not CD1a, S100 protein, CD34, or Mac387.629. and 633.
Mucinous histiocytosis. (A) There are epithelioid and spindle-cell histiocytes set in a collagenous and mucinous stroma. (B) This area is at the interface between the mucinous and more cellular areas seen in (A). (H & E)
Zebra and myeloid bodies are quite numerous in the cytoplasm of the histiocytes. This suggests a lysosomal storage phenomenon. Birbeck granules are absent.
Multicentric reticulohistiocytosis is an uncommon normolipemic histiocytosis that is characterized by the presence of an extensive papulonodular cutaneous eruption and a severe, sometimes destructive, arthropathy, the onset of which may precede, follow, or accompany the eruption of skin lesions.634.635.636.637. and 638. The interphalangeal joints of the hands are usually affected. Uncommonly, articular damage is minimal. 639 Oral, nasal, and pharyngeal mucosae are often involved. There are isolated reports of infiltrates in lymph nodes, lungs,640. and 641. bone marrow, the skull, 642 endocardium, stomach, salivary gland, 643 and perirenal fat. Xanthelasmas of the eyelids are also common. 635 Over 200 cases of this condition have now been described. A retrospective study of 96 of these patients was reported in 2001. 644
The cutaneous lesions, which preferentially involve the face and distal parts of the upper extremities, are multiple brown-yellow papules and nodules measuring 0.3–2 cm in diameter. Rare presentations have included the presence of lesions resembling neurofibromatosis645 or dermatomyositis, 646 and the localization of lesions to the sites of healed herpes zoster. 647 Onset of the disease is usually in middle life, and there is a predilection for females. Childhood onset is rare.648. and 649. The clinical course is variable, with eventual spontaneous regression of the skin lesions after 5–10 years or more. 650 There may be residual joint impairment as a consequence of macrophages in synovial fluid differentiating into osteoclasts and causing bone resorption. 651 Malignancies of various types may develop in up to 30% of cases.636.652.653.654. and 655. Myelofibrosis has also been associated with this condition. 656 Systemic vasculitis is rarely associated with multicentric reticulohistiocytosis.
Because of the rarity of the disease, no consistent approach to treatment has emerged. 657 A combination of an oral alkylating agent with prednisone has been used with variable success. 657 Because immunohistochemical staining of the infiltrate in this condition has demonstrated the presence of histiocytes of a monocyte–macrophage lineage, and an abundance of cytokines, including tumor necrosis factor-α (TNF-α), a trial of etanercept (an anti-TNF agent) has been used with prednisone. 658 Skin lesions and synovitis completely resolved with this regimen. 658 There is a single case report of success with alendronate. 457
A related entity is diffuse (multiple) cutaneous reticulohistiocytosis, in which multiple cutaneous lesions, identical histologically to those seen in multicentric reticulohistiocytosis, develop in the absence of arthritis or systemic lesions.659.660. and 661. The terms ‘reticulohistiocytoma’ and ‘reticulohistiocytic granuloma’ have been used for solitary cutaneous lesions of similar histology (see below).
There is a circumscribed non-encapsulated dermal and synovial infiltrate of mononuclear and multinucleate histiocytes with an eosinophilic, finely granular ‘ground-glass’ cytoplasm. The hallmark of the disease is the presence of the multinucleate cells, which measure 50 µm or more in diameter (Fig. 40.14). They have 3–10 or more nuclei, which may be placed haphazardly, or along the periphery, or clustered in the center of the giant cell. Mitoses are infrequent. Some giant cells may have foamy cytoplasm at the periphery. Phagocytosis of nuclear debris is uncommon. Transition from mononucleate to multinucleate forms can be seen. 663 Giant cells may be sparse in early lesions. The cytoplasm of the histiocytes contains PAS-positive diastase-resistant material, which usually stains with Sudan black as well.634. and 664. Reticulin fibers can be demonstrated around individual cells.
Multicentric reticulohistiocytosis. The characteristic multinucleate giant cells are scattered through the collagen of the dermis. (H & E)
Immunohistochemistry shows that the histiocytic cells express CD45, CD68, CD11b, vimentin, and HAM-56, but not S100 protein, Mac387, CD34, or CD1a.644.659.665. and 666. There are reports of the cells expressing factor XIIIa, but in one of these cases the cells also stained for S100 protein, suggesting an unusual lineage. 667
In addition to the histiocytes, small numbers of lymphocytes are scattered through the infiltrate. There may be some perivascular cuffing by lymphocytes in early lesions. 478 The walls of small blood vessels sometimes show ‘onion-skin’ thickening. 635
Epidermal changes are variable. There is sometimes thinning of the epidermis overlying the lesion, with loss of rete ridges; ulceration is uncommon. Usually there is a narrow grenz zone of uninvolved collagen which separates the undersurface of the epidermis from the tumor nodule below.
The cytoplasm of the histiocytes is rich in organelles. A characteristic feature is the presence of innumerable rounded dense bodies with the morphological structure of lysosomes. 663 Lipid vacuoles are sometimes present, but there are no Birbeck granules. 643 Elastin and collagen in various stages of degeneration may be seen in the cytoplasm of some of the giant cells.668. and 669. Intra- and extracytoplasmic long spacing collagen (type VI) has been reported in one case. 670
Reticulohistiocytomas (reticulohistiocytic granulomas) are nodular lesions 0.5–2 cm in diameter, with similar histology to the lesions of multicentric reticulohistiocytosis but without associated arthritis or systemic lesions.637. and 671. They are usually solitary, but several may be present.671.672. and 673. There is a predilection for the head and neck, but they may occur anywhere on the skin. Paraproteinemia accompanied several cutaneous nodules in one reported case. 672 In a report of 44 cases from the Armed Forces Institute of Pathology (AFIP), the term ‘solitary epithelioid histiocytoma’ was suggested as the preferable designation for this tumor. 674
There is a circumscribed dermal nodule, often with overlying epidermal thinning. As with multicentric reticulohistiocytosis, there is an irregular admixture of oncocytic mononuclear histiocytes, multinucleate giant cells with a ground-glass appearance, and inflammatory cells (Fig. 40.15). Phagocytosis of leukocytes is sometimes seen. A few Touton giant cells may be present and these may contain lipid. Reticulin fibers are increased and surround individual cells.
Solitary reticulohistiocytoma composed of multinucleate giant cells, histiocytes, and lymphocytes. There is some fibrosis of the stroma. (H & E)
There is usually more of a neutrophilic infiltrate than in multicentric reticulohistiocytosis, and a further point of distinction is the greater propensity for the stroma in reticulohistiocytoma to have many spindle-shaped cells and for there to be xanthomatized cells. 666
Immunohistochemistry suggests a dendritic cell lineage with staining for factor XIIIa, CD68, HAM-56, lysozyme, α1-antitrypsin, CD11b, and CD14. The cells are negative for CD1a, CD34, and smooth muscle actin.666.675. and 676. Positive staining for S100 protein has been reported in several cases, but it is usually negative.554. and 677. In the series reported from the AFIP (see above), the epithelioid histiocytes were positive for CD163, CD68, lysozyme (variably), and vimentin. 674
Familial histiocytic dermatoarthritis (OMIM 142730) is an exceedingly rare disorder that presents in childhood or adolescence as a papulonodular eruption on the face and limbs associated with a symmetrical destructive arthritis and ocular lesions. A closely related entity is the autosomal recessive dermo-chondro-corneal dystrophy (OMIM 221800) reported in the French literature. 678 The gene responsible has not yet been identified.
There is a dermal infiltrate of mononuclear histiocytes admixed with some lymphocytes and plasma cells. There is no stored lipid or PAS-positive cytoplasmic material. Fibrosis is conspicuous in older lesions. Two cases have been reported in which multinucleate histiocytes were present. PAS-positive material was present in the cytoplasm. The authors suggested that these two cases had features overlapping with both familial histiocytic dermatoarthritis and multicentric reticulohistiocytosis, which probably form part of a spectrum of dermatoarthritides. 678
Necrobiotic xanthogranuloma, a rare chronic disorder, was first delineated by Kossard and Winkelmann in 1980.679. and 680. It is characterized by the presence of multiple sharply demarcated nodules and large indurated plaques. 681 Sometimes only papulonodules are present. 682 A solitary variant has been described.683. and 684. Lesions are violaceous to red, with a partly xanthomatous hue. Central atrophy, ulceration, and telangiectasia may develop. There is a predilection for the periorbital area, but other areas of the face, as well as the trunk and limbs, are often involved.685. and 686. It has also been reported in a burn scar. 687
Paraproteinemia has been present in nearly all the reported cases.688.689.690.691.692. and 693. Two monoclonal paraproteins were present in one case. 694 Other less frequent findings include leukopenia, bone marrow plasmacytosis, non-Hodgkin’s lymphoma, 695 hypocomplementemia, 696 and, occasionally, hyperlipidemia. Normolipemic plane xanthoma has been reported in association with necrobiotic xanthogranuloma, suggesting that the two conditions are part of a spectrum.692. and 697. It has also developed within linear morphea. 698 Ophthalmic complications are common and include scleritis, episcleritis, and keratitis. Cardiac and laryngeal involvement has been recorded.699. and 700. Skeletal muscle has been involved without overlying dermal disease. 701 The clinical course is chronic and often progressive. 702
Necrobiotic xanthogranuloma has been regarded as a paraneoplastic phenomenon, but recent findings raise the possibility that it is an infectious disease in immunocompromised patients, in particular, in those with multiple myeloma. 703 This theory results from the finding of Borrelia organisms in six of seven cases using focus-floating microscopy, a technique in which sections are simultaneously scanned both horizontally and vertically. 703 As the authors point out it is possible that new strains of Borrelia or related spirochetes, not yet properly characterized could be involved in the etiology. 703
Treatment of necrobiotic xanthogranuloma is difficult owing to the chronic and progressive nature of the disease. Therapy is usually directed at the underlying hematological disorder. Melphalan, chlorambucil, methotrexate, cyclophosphamide, plasmapheresis, interferon alfa-2b, prednisone, and local corticosteroid injections have all been used. 693 Thalidomide has also been used to treat this disease. 704
The distinctive changes are found in both the dermis and subcutis, and consist of broad zones of hyaline necrobiosis and granulomatous foci composed of histiocytes, foam cells and multinucleate giant cells (Fig. 40.16). The giant cells are of both Touton and foreign-body type, the latter often having bizarre features with irregular size, shape, and distribution of the nuclei. Asteroid bodies and other inclusions are sometimes present. 706 The multinucleate cells may be present in the granulomas and dispersed near the zones of necrobiosis. Sometimes the Touton cells are prominent in the subcutis (‘Touton-cell panniculitis’). Giant cells are occasionally quite sparse. 707 Blood vessels may be secondarily involved in the granulomatous process.
(A) Necrobiotic xanthogranuloma. A zone of necrobiosis is surrounded by multinucleate giant cells and some histiocytes. (B) Cholesterol clefts and giant cells are present. (H & E)
The amount of xanthomatization is variable, and sometimes the foam cell population is small. 707 Aggregates of histiocytes may extend into the papillary dermis, and there may be ulceration. Other less constant changes include the presence of cholesterol clefts, 708 rarely with surrounding palisaded granuloma formation, the presence of lymphoid nodules, sometimes with germinal centers, and plasma cell collections. Eosinophils are rare. Transepithelial elimination of cholesterol crystals and cellular debris has also been documented. 689 The process is more cellular and has more atypical and prominent giant cells than necrobiosis lipoidica. In one case involving the shoulder region, necrobiosis (collagenolysis) was absent. 709
Histochemical findings include scant mucin in areas of necrobiosis, sparse or absent elastic fibers, focal lipid droplets in giant cells and histiocytes and in some areas of necrobiosis, and PAS-positive diastase-resistant granules in giant cells. The histiocytes stain with Mac387. Scattered cells are CD68 positive while only isolated cells stain for S100 protein. 707 In one report most dermal cells were CD68+, and there was focal positivity for CD10. 709
Electron microscopy has not contributed any useful further information.
Orbital xanthogranuloma is a rare cutaneous disease, first reported in 1991, and characterized by bilateral, rather symmetrical infiltrates around the eyes of adult patients.710.711. and 712. As such, it has some clinical resemblance to necrobiotic xanthogranuloma (see above). 713 There is no systemic involvement, but associated clinical diseases have included adult-onset asthma and hematological abnormalities. 712 Lesions enlarge very slowly over many years.
There is a diffuse dermal and subcutaneous infiltrate of histiocytes that range from spindle-shaped cells to oval cells, and to large cells with foamy cytoplasm. 712 They infiltrate the dermis and subcutis. Large cells with foamy cytoplasm and multiple nuclei with some resemblance to Touton cells are present. A variable admixture of lymphocytes, plasma cells, and eosinophils may be present. 712 Focal, mild necrobiosis (collagenolysis) has been noted indicating the close relationship of this disease to necrobiotic xanthogranuloma. 712
Only a few cases of cutaneous atypical histiocytosis have been reported.714. and 715. It is characterized clinically by the presence of one or more nodular lesions resembling cutaneous lymphoma. There has been a good response to various modes of treatment in the cases reported so far. The status of this entity is doubtful.
There is a monomorphous infiltrate of medium-sized histiocytoid cells with occasional cytoplasmic vacuoles and scattered mitoses.
The distinctive feature is the presence in the cells of giant multivesicular bodies and pleomorphic granules.
Indeterminate cells are a class of dendritic cells found in the dermis. They display similar histological and antigenic features to Langerhans cells: both express S100 protein and CD1a surface antigens. 716 They differ by lacking Birbeck granules and by possessing some histiocytic markers such as CD68.
Only a few cases of indeterminate cell histiocytosis have been reported. 582 They usually present as multiple reddish-brown to yellowish papules.717.718. and 719. A solitary congenital variant, 716 and a solitary acquired lesion on the mucosa of the glans penis have also been reported. 720 A large series of 18 patients was described in 2005. 721 The authors of this series speculated that this disease might represent various macrophage disorders identified at various time points in the inflammatory response. 721 A case that progressed to leukemia has been reported. 722 A case of indeterminate cell sarcoma has been reported. 723 Vinblastine has been used to treat aggressive variants. 724 Partial remission has been obtained using UVB phototherapy. 725 Electron beam therapy is another option. 726
The tumor is composed of a monomorphous infiltrate of mononuclear, and occasionally multinucleate, histiocytes intermingled with clusters of lymphocytes. The cytoplasm is pale and the nucleus is often clefted. 716 In one case the cells were spindled with interspersed epithelioid histiocytes. 719
The eponymous designation Rosai–Dorfman disease727 is more appropriate than ‘sinus histiocytosis with massive lymphadenopathy’, because it is now recognized that in some instances extranodal lesions, including lesions in the skin, may be the sole manifestation of this condition.728.729.730.731.732.733.734.735.736.737.738.739.740.741. and 742. Cutaneous Rosai–Dorfman disease appears to be a distinct entity with different age and race distributions from cases with nodal disease. 743 It also has a wider histopathological spectrum. 744 The typical presentation is with painless cervical lymphadenopathy, accompanied by fever, anemia, an elevated erythrocyte sedimentation rate and hypergammaglobulinemia, which has been polyclonal in all but one case.745.746. and 747. Any age may be affected, but 80% of cases develop in the first two decades of life. Extranodal lesions occur in almost one-third of cases, and the skin has been involved in more than 10% of the many cases now reported.728. and 746. Purely cutaneous disease is uncommon; over 100 cases have now been reported, most in the last few years.744.748. and 749.
Cutaneous lesions, which are usually multiple, have been varied in their clinical appearance. 728 There may be nodules up to 4 cm or more in diameter,750. and 751. papules which are erythematous or xanthomatous, 728 plaques, pustules, acneiform lesions, 752 pigmented macules, 729 and even a transient panniculitis. 753 In one case, the presentation mimicked a vasculitis. 754 The eyelids and the malar regions are the sites of predilection.729. and 755. In the study by Brenn and colleagues of purely cutaneous disease, there was no site predilection. 748 Furthermore, 13 of the 22 cases had multifocal disease. Only 4 of the 21 patients with pure cutaneous disease reported from Taiwan had multifocal involvement. 756
The disease usually runs a benign, albeit protracted, clinical course, sometimes with significant morbidity. 746 Death may sometimes result from infiltration of organs or from immunological disturbances.729.757.758. and 759. The lesions resolved, regardless of the treatment given in 6 of 13 patients, for whom follow-up data were available, in the series of 22 cases, referred to above. 748 It is a polyclonal disorder of macrophage origin. 456 Its association with coexisting Langerhans cell histiocytosis may represent a reactive proliferation of Langerhans cells within a lesion of Rosai–Dorfman disease.749. and 760. Its association with other histiocytoses may have implications for its pathogenesis. 744 The occurrence of other malignancies is so rare that it may be incidental. 761 Human herpesvirus-6 (HHV-6) has been isolated from a skin lesion in one case. 762 Both HHV-6 and HHV-8 were not detected in a further three cases. 763 Recently large numbers of cells positive for parvovirus B19 were found by immunohistochemistry in four cases, suggesting that this may be the etiological agent. 764
When anatomically feasible, complete excision is the treatment of choice for persistent or recurrent extranodal disease.749. and 765. When surgical margins are involved, local recurrence is likely. Radiotherapy has shown limited efficacy, while chemotherapy is generally ineffective. 765 Isotretinoin has been used to treat this condition,755. and 766. while high-dose thalidomide has been used for extensive cutaneous disease. 756 Corticosteroids were used in another case. 767 Dapsone has been suggested for cases with a neutrophilic predominance. 768 Recently the tyrosine kinase inhibitor imatinib was used successfully to treat systemic disease. 476
The pathological changes in the lymph nodes are characteristic, and include expansion of the sinuses by large foamy histiocytes admixed with plasma cells. Cutaneous lesions show a dense dermal infiltrate of large polygonal histiocytes admixed with inflammatory cells. The histiocytes have abundant, lightly eosinophilic cytoplasm and vesicular nuclei. 728 These cells show variable emperipolesis (intracytoplasmic inflammatory cells).748. and 756. Scattered multinucleate cells and Touton cells and collections of neutrophils may be present.744. and 745. Plasma cells are invariably present, and sometimes contain prominent Russell bodies. Nodular lymphoid aggregates may be conspicuous.
Other features which may be present include xanthoma cells,749. and 769. fibrosis, increased vascularity, and focal necrosis. 728 In some lesions there is a markedly sclerotic, often storiform, fibrous stromal response. 748 Rarely the pattern is that of an inflammatory pseudotumor. 770 Histiocytes are often present in dilated lymphatics in the dermis. 735 A mixed septal and lobular panniculitis with some features of cytophagic histiocytic panniculitis (see p. 466) was present in one case. 753 Conspicuous rhomboidal and needle-shaped crystals were present in the cytoplasm of many lesional plasma cells and histiocytes in one case. Extracellular deposition was also present. 771
The large histiocytes of Rosai–Dorfman disease are S100 positive and negative for CD1a.730.772. and 773. They also express various macrophage and monocyte markers such as α1-antitrypsin, lysozyme, CD14, CD68, stabilin-1 (a marker of non-Langerhans cell histiocytes), 476 and Mac387.734. and 774. Expression of factor XIIIa, a dendrocyte marker, has also been reported775 but it has been negative in other cases as might be expected from its presumed cell of origin. 769 There are no Birbeck (Langerhans) inclusions in the histiocytes. 730
While it is most unusual to create an entity, in a textbook, on the basis of a solitary report, the case described with this designation seems sufficiently unique to justify a separate description. 776 The patient, with history of thrombocytosis, presented with a 5-year history of a violaceous, maculopapular rash primarily on her legs. The rash subsequently progressed to form confluent patches and plaques on her torso and arms. The clinical impression was disseminated granuloma annulare. 776
There was an interstitial dermatitis and a mixed lobular and septal panniculitis with focal lymphoid aggregates. 776 The interstitial histiocytes had large, irregular nuclei with prominent nucleoli. Scattered, often bizarre multinucleate giant cells were present in the dermis, but concentrated near the dermal–subcutis interface. Focal aggregates of mature lymphocytes were present. There were no granulomas or necrobiosis. 776
The cells expressed CD68 and S100, but not CD1a.
This rare, life-threatening histiocytosis is characterized by a benign proliferation of histiocytes and an uncontrolled phagocytic syndrome, induced by T-cell activation, which in turn may be due to infections of various types. 457 Epstein–Barr virus is sometimes the triggering infection. 777 There may be an underlying connective tissue disease, malignancy, or HIV infection that depresses the immune system. 778
Patients present with fever, splenomegaly, liver dysfunction, cytopenia, hypofibrinogenemia, and tissue hemophagocytosis. 456 Cutaneous manifestations are present in up to 65% of cases, but they are a minor component of the illness. 779 There may be hemorrhage from the hypofibrinogenemia.
A dysfunction of the cytokine perforin seems to be involved in many cases. Its release may be triggered by the precipitating infection. 457 Perforin is implicated in the mechanism of at least two of the four familial hemophagocytic lymphohistiocytosis (HPLH) syndromes. These syndromes have predominantly been reported from parts of Europe, including Turkey, and from Japan. They are listed in Table 40.8. Hemophagocytosis can also be seen in cytophagic histiocytic panniculitis. The median survival of untreated cases is 2–3 months. Treatment of the triggering infection is the first requirement. Ultimately, hematopoietic stem cell transplantation may be required; it is curative. 457
There is usually non-specific spongiosis and a mild perivascular infiltrate of lymphocytes and histiocytes. 457 There is often focal hemorrhage. The histiocytes in the skin are S100 negative. Some are CD68 positive.
Histiocytic sarcoma is the preferred term of the International Lymphoma Study Group for a rare malignant tumor of histiocytes that was formerly called histiocytic medullary reticulosis, and later malignant histiocytosis. 780 Many of the earlier cases were not histiocytic when archival material has been subject to current immunohistochemical markers. 780 A review of seven suspected cases seen at the Mayo Clinic over a 20-year period produced only one case that had a true histiocytic origin. 781 Fletcher and colleagues reported 14 cases in 2004. 782 Clinical features include fever, generalized lymphadenopathy, hepatosplenomegaly, pancytopenia, and sometimes disseminated intravascular coagulation, night sweats, and abdominal pain.783.784. and 785. The skin is involved in 10–15% of cases, and in some this is the presenting feature.786.787. and 788. Lesions take the form of papules, nodules, or plaques anywhere on the body, but with a predilection for the extremities and buttocks. 789 Skin involvement mimicked kwashiorkor in one case. 790 Sometimes only a solitary nodule is present initially. Spontaneous healing of individual lesions may occur.
Malignant histiocytosis occurs at all ages; some cases have been reported in childhood.791.792. and 793. As some of these cases may have been other diseases, it is difficult to be certain of the percentage of childhood cases. The prognosis at all ages is poor, although patients presenting with skin lesions form a subgroup with a more favorable outlook.783. and 786. Prognosis might be related to tumor size. 782 At autopsy there is usually involvement of many organs. In the series of 18 cases reported by the International Lymphoma Study Group, the median age of the patients was 46 years; 72% presented with extranodal disease. The mortality rate was 58%. 780
Polytherapy consisting of vincristine, cyclophosphamide, doxorubicin, and prednisolone achieved complete remission in 22 out of 27 patients in one series. 457
Two patterns of cutaneous involvement may be seen. 787 In one there is a dense, predominantly periadnexal and perivascular infiltrate of atypical histiocytes; in the other there is a diffuse infiltrate of similar cells in the deep dermis and subcutis, with focal necrosis. The papillary dermis is usually spared, but if there are cells in this region there is no associated epidermotropism such as is seen in Langerhans cell histiocytosis. 794
The cells in the infiltrate show variable pleomorphism, which increases with the passage of time. The cells measure 15–25 µm in diameter and have an oval or reniform nucleus and abundant eosinophilic cytoplasm. Occasional binucleate cells are present. Phagocytosis of erythrocytes and nuclear debris may be seen, but it is not a universal feature. 789 There are also some small lymphocytes and plasma cells in the infiltrate. 794
The nuclear atypia in malignant histiocytosis allows it to be distinguished from the virus-associated hemophagocytic syndrome, histiocytosis X, and cytophagic panniculitis. 786
In the series of 18 cases, reported by the International Lymphoma Study Group and referred to above, the following phenotype was expressed: CD68 (100%), lysozyme (94%), CD1a (0%), S100 (33%), CD21/35 (0%). 780 Concavalin-A is also present. 784 CD31 may be a marker for some histiocytic malignancies. 795 A case of histiocytic sarcoma of the peritonsillar region but with cutaneous lesions with the immunophenotype of Langerhans cells (CD1a+, S100+) but without Birbeck granules (that is, indeterminate cells) has been reported. 796
The term ‘reactive histiocytosis’, also known as secondary histiocytosis, refers to the increased number of histiocytes that may be seen in a variety of cutaneous infections. These include histoplasmosis, toxoplasmosis, brucellosis, tuberculosis, leprosy, rubella, and certain infections with the Epstein–Barr virus. 450 Histiocytes are also prominent in actinic reticuloid. Histiocyte-like myeloid cells have been reported in some cases of Sweet’s syndrome (histiocytoid Sweet’s syndrome). 799 In susceptible hosts, particularly those with various immunodeficiency syndromes, infections may trigger idiopathic histiocytic proliferations indistinguishable from Langerhans cell histiocytosis. 450
A histiocytic infiltrate has also been reported in two patients with acute lymphocytic leukemia receiving cytarabine. 800 Lesions resolved after cessation of the drug.
Intravascular/intralymphatic histiocytosis is a rare, reactive histiocytosis characterized by the proliferation of histiocytes in vascular or lymphatic lumina.801.802. and 803. Most of the reported cases have been associated with rheumatoid arthritis,804.805.806. and 807. but tonsillitis was the associated condition in another patient. 803 Intralymphatic histiocytosis has been reported in the region of the metal implants of a knee joint replacement. 808 Granulomas were also present. Histiocytes are sometimes seen in dilated lymphatics in the dermis in Rosai–Dorfman disease. 735
Lesions associated with rheumatoid arthritis are usually asymptomatic, irregularly shaped patches of erythema, sometimes livedo-like, in the vicinity of the involved joint. The case associated with tonsillitis presented with painful induration of the scrotum. 803
There are heavy intralesional aggregates composed mainly of histiocytes within dilated vessels in the dermis. Thrombosis of vessels was present in the scrotal case (see above). 803 There is a mild, but variable infiltrate of lymphocytes, plasma cells, and sometimes neutrophils in the intervening dermis. 809 In one report, the intraluminal cells expressed CD68, Mac387, and CD43, but not CD79a, CD20, CD3, or vascular endothelial markers such as CD31 and factor VIII. 803 The endothelial cells expressed vascular markers, but not D2-40, a marker for lymphatic endothelial cells. 803 However, in three other reports, the intraluminal cells also expressed CD68, but the endothelial cells expressed the lymphatic marker D2-40.804.805. and 808.
Xanthomas represent the accumulation of lipid-rich macrophages known as foam cells. 810 They present clinically as yellow or yellow-brown papules, nodules, or plaques, the color depending on the amount of lipid present and its depth below the surface. They are usually associated with disorders of lipoprotein metabolism, although only a minority of individuals with such disorders develop xanthomas.810. and 811.
Xanthomas can be further subdivided on the basis of their clinical morphology, anatomical distribution, and mode of development into the following types: eruptive, tuberous, tendinous, planar, verruciform, and papular.810. and 811. In addition, there are isolated reports in the literature of xanthomas which cannot be fitted into an orderly classification.812.813.814.815.816. and 817. One such example is the occurrence of normolipemic xanthomas in association with HIV infection.818. and 819. Another is the solitary xanthoma with prominent epidermotropism simulating balloon cell melanoma. 820 This latter case may have been a fibrous papule with clear cells and not a xanthoma. It is difficult to explain the occurrence of multiple xanthomas on the ears of a normolipemic adolescent. 821 Specifically excluded, perhaps arbitrarily so, are the xanthogranulomas (which are not related to disorders of lipoprotein metabolism and which usually possess an admixture of cell types), and the histiocytic, dendrocytic, and Langerhans cell proliferations in which lipid accumulation (xanthomatization) may be a secondary phenomenon.811.813. and 822. Tangier disease and disseminated lipogranulomatosis (Farber’s disease), which also possess some xanthoma cells, are not usually included with the xanthomas. The various diseases featuring the presence of xanthoma cells in the skin are summarized in Table 40.9.
Several of the xanthoma types mentioned above are associated with specific abnormalities of lipoprotein metabolism; more than one type of xanthoma may be present in a particular lipoprotein disorder.811. and 823. The lipids in xanthomas are primarily free and esterified cholesterol, but occasionally other sterols and even triglycerides accumulate. This is usually the result of a high plasma concentration with subsequent permeation of lipoproteins through the walls of dermal capillaries.810. and 824. The lipid is then taken up by dermal macrophages, which evolve into foam cells.810. and 824.
Attention has been given recently to the mechanism by which these monocyte-derived macrophages accumulate in the skin. The process is similar to atherosclerosis and involves vascular endothelial cells and adhesion molecule expression. 825 The presence of increased numbers of E-selectin-positive endothelial cells and a decrease in ICAM-1 cells promotes macrophage migration into xanthoma lesions. 825
There are several possible explanations for the formation of xanthomas in normolipemic states. 824 There may be altered lipoprotein content or structure, or an underlying lymphoproliferative disease with xanthomatization of cells infiltrating the dermis. 824 Finally, local tissue factors may play a role in those cases of xanthoma developing in chronic eczema, photosensitive eruptions, erythroderma, and lymphedema, and at sites of injury such as bites, scars, and striae and in some regressing tumors. The term ‘dystrophic xanthoma’ has been used for the accumulation of lipid-rich foam cells within an area of abnormal or damaged skin in both normolipemic and hyperlipoproteinemic states.826. and 827. Dystrophic xanthomas may develop in burns scars, lymphedema praecox, 828 laparotomy scars, striae, lichen planus, 829 mycosis fungoides, and at the sites of mosquito bites, vaccination, herpes zoster, and phlebitis.
The various types of xanthoma will now be considered.
In the eruptive variant of xanthoma, multiple small red-yellow papules with an erythematous halo develop in crops. 810 There is a predilection for the buttocks and thighs, and the extensor surfaces of the arms and legs. 811 The lesions become progressively more yellow and eventually resolve spontaneously over several weeks.
Eruptive xanthomas occur in a setting of elevated plasma chylomicrons such as may occur in uncontrolled diabetes mellitus or following the ingestion of alcohol or the use of exogenous estrogens.811. and 830. Rare associations include lipoprotein lipase deficiency, 811 types IV and V hyperlipoproteinemia, 831 normolipemia, 832 pregnancy, 833 the nephrotic syndrome, 834 chylous effusions, 835 hypothyroidism, the use of intravenous miconazole, 836 and the oral ingestion of 13-cis-retinoic acid. 837 Eruptive xanthomas have followed the use of the antipsychotic drug olanzapine838 and the protease inhibitor ritonavir. 839
The unique case of normolipemic eruptive xanthomas associated with generalized edema is best regarded as a variant of dystrophic xanthomatosis (see above). 840
The architecture of the reticular dermis is disturbed by an infiltrate of cells and extravascular lipid deposits in the form of lace-like eosinophilic material between the collagen bundles. The cellular infiltrate cuffs capillaries and extends throughout the dermis. 841 Initially it is composed of neutrophils and lymphocytes, and histiocytes with a finely stippled cytoplasm. 841 Neutrophils may be quite prominent in early lesions, and the absence of foam cells makes the diagnosis difficult on histopathological grounds alone. In established lesions the lipidization of cells is more obvious, although foam cells are never as obvious as in the other variants of xanthoma (Fig. 40.17). 842
Eruptive xanthoma. Small foam cells are admixed with lymphocytes, macrophages, and neutrophils. (H & E)
In some stages of their evolution eruptive xanthomas may mimic the changes seen in granuloma annulare, but close inspection for the features mentioned above allows the distinction to be made. 841 Furthermore, doubly-refractile lipid can be seen between foam cells in formalin-fixed material in some cases of eruptive xanthoma, but not in granuloma annulare. 843 In one report, the doubly refractile material was said to resemble urate crystals. 844
Tuberous xanthomas present a spectrum which ranges from small inflammatory lesions at one end (tuberoeruptive xanthomas) to large nodular lesions at the other. Tuberous xanthomas often form by coalescence of smaller lesions. 810 They are yellowish in color and are found particularly on the elbows, knees, and buttocks. With treatment of the underlying hyperlipidemia there is usually slow resolution over many months.
Tuberous xanthomas are most characteristic of familial hyperlipoproteinemia (type III), but they can be seen rarely in homozygous and heterozygous familial hypercholesterolemia, 845 which includes autosomal recessive hypercholesterolemia (OMIM 603813) due to mutations in the ARH gene at 1p36–p35, 846 hepatic cholestasis,847.848. and 849. cerebrotendinous xanthoma, and β-sitosterolemia. 811 This latter condition (OMIM 210250) is autosomal recessive, with the gene localized to 2p21. 850 It is caused by mutations in the ABC68 gene or in the ABC65 gene, both of which are located at this gene locus. 851 Tendinous xanthomas are also present in these latter two conditions, as well as in the hyperlipidemia associated with the protease inhibitor ritonavir. 852 Tuberous xanthomas have also been reported in a normolipidemic subject. 853
There are large aggregates of foam cells throughout the dermis, but usually no Touton giant cells or other inflammatory cells (Fig. 40.18). Fibroblasts are increased in number in older lesions, leading to the progressive deposition of collagen. 853 In one case, concentric layers of xanthoma cells surrounded a cutaneous nerve. 854 In another the cells assumed a plexiform pattern – plexiform xanthoma.855. and 856.
Xanthoma tuberosum. (A) Sheets of foam cells involve the dermis. (B) Another case. It is early and the xanthoma cells are much smaller than in lesions of long standing. (H & E)
The lipid within the foam cells can be stained with oil-red O in frozen sections, or examined polariscopically to confirm its doubly refractile property.
In tendinous xanthoma, lesions of varying size develop in ligaments, fasciae, and tendons, especially the extensor tendons of the hands and feet and the Achilles tendon. 811 They are firm to hard, flesh-colored nodules which develop slowly over decades.
Tendinous xanthomas are most commonly associated with heterozygous familial hypercholesterolemia, but they have also been reported in cerebrotendinous xanthoma (caused by a mutation in the sterol 27-hydroxylase gene), β-sitosterolemia, familial hyperlipoproteinemia (type III) and hepatic cholestasis, and rarely in normolipidemic individuals.811.853. and 857. Cerebrotendinous xanthomatosis (OMIM 213700) is caused by a mutation in the CYP27A1 gene, which encodes sterol 27-hydroxylase, on chromosome 2q33–qter.
The appearances are similar to those seen in tuberous xanthomas, except for the different tissue substrate.
Plexiform xanthomatous tumor is a normolipemic xanthomatous tumor with a distinctive plexiform pattern, composed of a variable admixture of uniform epithelioid and xanthomatous cells. 858 Lesions are usually solitary, but up to four lesions may be present. All cases reported have been in males. 858 Some cases may represent a morphological variant of tuberous or tendinous xanthoma, but lesions are usually smaller than those in these other two variants of xanthoma.
The tumors are located in the dermis and subcutis. As mentioned above, there is a distinctive plexiform arrangement of epithelioid and xanthomatous cells. Rarely cholesterol clefts, necrosis, and Touton giant cells are present. There are sparse inflammatory cells. The cells stain with CD68, but are negative for S100, CD34, and cytokeratins. 858
Planar xanthomas are yellow, soft macules or slightly elevated plaques which are further subdivided on the basis of their location into xanthelasmas, intertriginous xanthomas, 811 xanthoma striatum palmaris,831. and 859. and diffuse (generalized) plane xanthomas.810. and 811. A further variant, planar xanthoma of cholestasis, is sometimes recognized.811. and 860.
Xanthelasma is the best known and most common form of xanthoma and is characterized by one or more yellowish plaques on the eyelids or in periorbital skin. 861 In one patient, the xanthelasma was unilateral, sparing a paralyzed lid. 862 Lipid levels are normal in approximately 50% of affected individuals, although in young affected persons there is a higher incidence of hypercholesterolemia.810.848. and 863. Abnormalities in the apoprotein moiety of lipoproteins have been detected in some of those with normal levels of lipid in the blood.864.865. and 866. Altered vascular permeability may also play a role. 867 Its association with the hyperimmunoglobulinemia syndrome combined with a lymphoma was probably fortuitous. 868
Many treatments have been used to remove these unsightly lesions. They include surgical resection, trichloroacetic acid peeling, and treatment with various types of laser, including CO2 lasers, erbium:YAG laser, or pulsed dye laser. 869 The 1064 nm Q-switched Nd:YAG laser is another treatment option. 869
The rare condition of diffuse (generalized) plane xanthomas is associated with macular, yellowish discoloration of the skin, involving particularly the trunk and neck, and sometimes the face.867.872. and 873. Yellow-orange plaques are sometimes present. 874 It usually occurs in adults; childhood cases are rare. 875 The disease usually runs a protracted course. The majority of patients are normolipidemic, although several hyperlipoproteinemic states have been associated with this variant. 876 Increased vascular permeability is sometimes present. 867 There is a significant association with lymphoreticular neoplasms, particularly myeloma, although these disorders may not become manifest until several years after the onset of the xanthomas.877.878.879.880.881.882. and 883. Other associations include adult T-cell lymphoma/leukemia, 884 photosensitivity, 885 and adenocarcinoma of the rectum. 886 Sometimes paraproteinemia, without myeloma, is present. 874 Transformation into necrobiotic xanthogranuloma has been reported. 692
In xanthelasmas there are small aggregates of foam cells in the upper dermis. There is no fibrosis, and no other inflammatory cells are present except a mild increase in mast cells. 887 Plane xanthomas are composed of small groups and streaks of foam cells in the upper dermis, and sometimes around pilosebaceous follicles. A few perivascular lymphocytes are sometimes present. Fibroblasts may also be increased in number. Uncommonly, there is a more diffuse infiltrate of foamy macrophages and Touton cells, with cholesterol clefts and necrobiosis resembling the pattern seen in necrobiotic xanthogranuloma. 874
Electron microscopy confirms the transition of macrophages into foam cells with lipid inclusions in the cytoplasm.
The verruciform variant of xanthoma is composed of asymptomatic, usually solitary, flat plaques or warty lesions up to 2 cm in diameter. 888 A giant lesion measuring 15 × 20 cm has been reported. 889 They may vary in color from gray to pink or yellow, depending on the thickness of the overlying epithelium. There is a marked predilection for the oral cavity, 890 although lesions have also been reported on the vulva, 891 perianal skin, 892 scrotum,893.894. and 895. penis, 896 and occasionally extragenital skin.897.898. and 899. Multiple papules of the hands and feet have been reported in association with verrucous genital lesions. 900 Verruciform xanthomas have developed in association with epidermal nevi,901. and 902. a seborrheic keratosis, 903 a fibroepithelial polyp of the vulva, 904 a squamous cell carcinoma,905. and 906. an arteriovenous hemangioma, 907 recessive dystrophic epidermolysis bullosa,898. and 908. discoid lupus erythematosus, 909 and lymphedema of the leg.910.911.912. and 913. They have occurred in immunocompromised patients with HIV-1 infection, 914 with graft-versus-host disease, 915 and in association with human papillomavirus (HPV).916.917. and 918. Because similar morphological changes are identified in CHILD syndrome (see p. 256) resulting from mutational inactivation of the 3β-hydroxysteroid dehydrogenase (NSDHL) gene, located at Xq28, a search was made for mutations in this gene in cases of sporadic verruciform xanthoma. A novel missense mutation in exon 6 of the NSDHL gene was demonstrated in two of the nine patients tested. 919 This mutation is not the usual one encountered in the CHILD syndrome. Lesions may persist for long periods. It is assumed that the formation of the xanthoma cells is secondary to degeneration of or damage to cells in the overlying epithelium.897.914.920.921. and 922. The xanthoma cells are thought to be derived from dermal dendrocytes. 923 Lipid metabolism is usually normal, although verruciform xanthoma has been reported in association with an undefined systemic lipid storage disorder. 924
On low-power magnification the lesions often have a verruca-like configuration. There is hyperkeratosis, focal parakeratosis, and verrucous acanthosis (Fig. 40.19). Keratinous columns sometimes extend down into invaginations of the epidermis. There is often prominent exocytosis of neutrophils into the upper layers of the epithelium and the parakeratotic scale. 925 The basal layer usually has reduced amounts of melanin. Cystic invagination of the surface epidermis has been reported. 926
Verruciform xanthoma. (A) There is marked hyperkeratosis and verrucous acanthosis, with numerous foam cells in the papillary dermis. (B) The foam cells show some nuclear variability. (H & E)
The papillary dermis is filled with numerous large xanthoma cells. There are variable numbers of lymphocytes, plasma cells, neutrophils, and eosinophils beneath and between the xanthoma cells. The cells contain lipid and small amounts of PAS-positive diastase-resistant material between the lipid vacuoles. Perivascular hyalinization of superficial vessels is sometimes present. Perifollicular deposits have been reported in patients with clinical features of Fox–Fordyce disease. 927
The foam cells are positive for CD68 and weakly positive for cytokeratin and factor XIIIa, but negative for S100 protein. 904
Papular xanthoma is a very rare condition consisting of multiple discrete yellow-red papules on the face and trunk and occasionally on mucous membranes.449. and 478. Only eight cases have been reported, according to a paper in 2007. 929 The condition has been classified with the histiocytic infiltrates because affected individuals are usually normolipidemic, 449 but as there are no primitive histiocytes or other inflammatory cells in the lesions it seems best to classify it as a xanthoma. 930 Furthermore, qualitative abnormalities in the lipoproteins may be present. 931 Papular xanthoma, particularly in children, may be clinically indistinguishable from juvenile xanthogranuloma. 932 Rarely, it is congenital. 933
Papular xanthomatosis has been reported in a patient with the Sézary syndrome, 934 and in one with erythrodermic atopic dermatitis. 930
The skin lesions in adults tend to be persistent, although childhood cases are often self-healing within 1 to 5 years. 933 An adult case has been treated successfully with doxycycline. 929
There is an infiltrate of foam cells in the upper and mid dermis, with variable numbers of Touton giant cells. There are very few, if any, chronic inflammatory cells within the lesions. Small amounts of hemosiderin are sometimes present within the foam cells and in extracellular locations. The cells are S100 negative. In one reported case the foam cells stained strongly for factor XIIIa, but were negative for Mac387. However, the multinucleate cells expressed this latter marker. 936 In another study, the cells were positive for CD68, but negative for S100 protein, CD1a, CD56, and factor XIIIa. The authors concluded that the cells were of macrophage rather than dendrocyte origin. 937 A similar finding was reported in the large series of 10 cases by Breier and colleagues938 who noted reactivity for macrophage markers such as KiM1p, CD68, and HAM-56. 938 The opposite conclusion was expressed in a recent paper, in which the cells expressed CD68 and factor XIIIa, but not CD1a and S100, typical of a dendrocyte origin. 929
The absence of an inflammatory cell component distinguishes papular xanthoma from xanthoma disseminatum, eruptive xanthoma, and juvenile xanthogranuloma, and clinical features allow its distinction from the other xanthomas.
The foam cells are macrophages with numerous lipid vacuoles and some myelin bodies. 935
There is one report of three patients with HIV infection who developed a papular facial eruption in association with an IgA gammopathy. 819 Although there is some clinical similarity with the plane or papular xanthomas associated with lymphoproliferative disorders, the histology of these lesions is unique.
The epidermis is intact with a dermal infiltrate of xanthoma cells. Between the xanthoma cells there is hyalinized collagen with neutrophils and nuclear dust. Some of the foamy macrophages contain nuclear debris within their cytoplasm. 819
The Langerhans cell is one of the dendritic cells, a group of non-phagocytic mononuclear cells which play a role in the trapping and processing of antigens for presentation to lymphocytes.939. and 940. Dendritic cells may be of mesenchymal or hemopoietic stem cell origin.456. and 941. Other dendritic cells include so-called ‘indeterminate cells’, ‘interdigitating dendritic (reticulum) cells’, 942‘follicular dendritic cells’, and ‘veiled cells’. Tumors of these other dendritic cells are exceedingly rare, with only a few cases reported.943.944.945.946. and 947.
Epidermal Langerhans cells are derived from bone marrow. 457 They account for approximately 2% of the cells in the epidermis. 939 Like other dendritic cells they specialize in processing and presenting antigens to T lymphocytes. 948 Epidermal Langerhans cells are particularly efficient in processing bacterial antigens.949. and 950. As Langerhans cells move from the skin and travel to draining lymph nodes, they lose their ability to process antigen and acquire the characteristics necessary to present antigen to T lymphocytes. 951 In the lymph nodes, the Langerhans cells may be the cells that have been referred to as veiled cells. 941 Various epidermal cytokines facilitate this functional maturation including granulocyte–macrophage colony-stimulating factor (GM-CSF), TNF-α and interleukin-1 (IL-1). This mechanism ensures that antigen presenting at the skin surface is translated into an appropriate immune response. 951 The migratory response of epidermal Langerhans cells to TNF-α decreases in the elderly. 952 They are also reduced in number with age and there is atrophic morphology with fewer dendrites, and fewer Birbeck granules. 953
Langerhans cells are approximately 12–15 µm in diameter with clear cytoplasm and an irregularly shaped nucleus. They are situated in the epidermis above the basal layer, a situation that usually distinguishes them from melanocytes in hematoxylin and eosin-stained sections and in immunoperoxidase preparations for S100 protein. There is considerable regional variation in the number of Langerhans cells in the epidermis.954. and 955. Langerhans cells are also found in the pilosebaceous apparatus, where they localize to the infundibular region of the follicle with extension into the germinative sebaceous epithelium. 956
Ultrastructurally, Langerhans cells possess many organelles, the most characteristic of which is the Birbeck (Langerhans) granule. This is a rod or tennis racquet-shaped organelle of variable length and a constant width of 33 nm. 939 Similar dendritic cells without Birbeck granules are known as indeterminate cells. 939 Of interest is the report of a healthy white male whose epidermal Langerhans cells lacked Birbeck granules. The cells appeared to have normal antigen-presenting capacity, suggesting that Birbeck granules are not a prerequisite for normal function. 957
Langerhans cells show esterase, acid phosphatase, and ATPase activity. 958 They express certain antigens such as CD1a, S100 protein, CD45, CD101, and HLA-DR, and they bind peanut lectin.958.959. and 960. CD1a staining is considered specific for Langerhans cells. Birbeck granules are recognized by the monoclonal antibody anti-Lag (Langerhans-associated granule), but another antigen langerin is also detected with anti-Lag.457. and 961. Langerin (CD207) acts as an endocytic receptor to translocate ligand from the cell surface into the Birbeck granule. This suggests a functional importance for the Birbeck granule as a novel pathway for antigen processing. 962 Recent work suggests that the dermis contains dendritic cells that are langerin+, which function independently of epidermal Langerhans cells. 963 Although epidermal Langerhans cells do not express fascin, a dendritic cell marker in lymphoid tissue and peripheral blood, it is found in the cells of Langerhans cell histiocytosis. 964
Langerhans cells play an important role in the pathogenesis of allergic contact dermatitis, and a lesser role in various other inflammatory dermatoses, such as lichen planus. 939 They have also been seen in the nodules in scabies, as have indeterminate cells. 965 Langerhans cells are often increased in number in the epidermis overlying various cutaneous tumors. 966 Flask-shaped pseudo-Pautrier abscesses (Langerhans cell microgranulomas) are sometimes present in spongiotic dermatitides, imparting a pityriasiform appearance. 967 They are sometimes present in allergic contact dermatitis. They possibly result from accelerated migration and activation from peripheral blood and dermal precursors. 967 However, significant infiltrates of Langerhans cells are found only in Langerhans cell histiocytosis (formerly known as histiocytosis X) and congenital self-healing histiocytosis, which is now regarded as a self-limited variant of Langerhans cell histiocytosis. 968 The reduction in Langerhans cell numbers in the vulva in HIV-positive patients, lowers local vulvar immunity and may contribute to the progression of HPV-related vulvar intraepithelial neoplasia. 969 Accumulating evidence indicates that anti-inflammatory drugs target dendritic cells on multiple levels, including maturation, migration, and differentiation. 970