Fig. 32.1
Potential sources of myofibroblasts. Myofibroblasts, besides resulting from the differentiation of normal cutaneous resident fibroblasts, may also derive from several cellular sources, including pericytes, epithelial cells, endothelial cells, circulating fibrocytes, and mesenchymal stem cells. EMT epithelial to mesenchymal transition, EndMT endothelial to mesenchymal transition (Figure adapted per Creative Commons license from Micallef et al. [253])
Furthermore, growing evidence suggests that subcutaneous adipocyte progenitors may also contribute to fibrosis, especially in SSc, in which adipose tissue is replaced by fibrotic tissue [19, 20]. Finally, both epithelial cells and endothelial cells have been postulated to trans-differentiate into fibroblasts in fibrosing skin and other tissues. Until further studies dissect the relative contributions of these diverse lineages to different fibrotic conditions, each of these cell types can be considered a potential source of matrix-producing fibroblasts in addition to the resident dermal fibroblasts.
Endothelial Cells
Endothelial cells are crucial mediators of the wound healing process (see Section Cutaneous Wound Healing). In fibrotic skin disorders, endothelial or microvascular dysfunction may play an important role in early stages of injury response leading to fibrogenesis [21].
The role of endothelial cells in fibrosis is of particular interest in SSc (see Section Scleroderma/Systemic Sclerosis), which is accompanied by skin microvessel vasculopathy and endothelial apoptosis and is often preceded by the appearance of Raynaud’s phenomenon [22]. Tight-skin mice, which have an in-frame duplication of one allele of the gene encoding fibrillin-1, recapitulate several features of SSc, including excess collagen, thickened skin, and endothelial dysfunction [23]. In addition to contributing to tissue hypoxia, endothelial cell dysregulation may also directly contribute to fibrosis via endothelial-to-mesenchymal transition (EndMT), a phenomenon that produces activated fibroblasts in injured tissue [24, 25].
How endothelial cells might contribute to fibrotic scar formation is unclear. In contrast to SSc, keloidal and hypertrophic scars do not exhibit endothelial cell apoptosis [26]. Instead, dermal microvessels are more abundant in these scar tissues than in normal skin and tend to exhibit occlusion that may contribute to local tissue hypoxia [27, 28].
Epithelial Cells
Epithelial cells, including basal keratinocytes and hair follicle-associated stem cells, contribute to re-epithelialization during wound healing [2]. Therefore, in wound healing and scar formation, epithelial proliferation and migration are key steps that regulate fibrotic processes (see Section Cutaneous Wound Healing). As discussed in Section Activated Fibroblasts and Myofibroblasts, epithelial-to-mesenchymal transition (EMT) may serve as a source of matrix producing cells. EMT, a phenomenon in which epithelial cells transdifferentiate into mesenchymal cells, has important roles in embryonic development and tumorigenesis. It has been suggested that EMT contributes to the activated fibroblast population in fibrosis of the kidney, lung, and liver [29]. In contrast, the role of EMT in skin fibrosis has not been carefully studied [30]. In a transgenic mouse model of fibrosis, overexpression of profibrotic CTGF in mouse fibroblasts in vivo was sufficient for an EMT gene expression signature to become detectable in skin and lung epithelium [31]. However, dedicated lineage-tracing studies are still required in order to demonstrate a role for EMT in the bleomycin-induced and tight-skin murine models of skin fibrosis.
T Lymphocytes
T cells are present in the inflammatory phase of normal wound healing as well as in keloidal scars and lesional tissue affected by SSc and acute graft versus host disease (GVHD) [2, 32–34]. T lymphocytes have several potential roles in fibrosis. First, they can stimulate fibroblasts to produce excess collagen [35]. Second, they can induce local macrophages to produce profibrotic cytokines [21]. Finally, they can promote vascular dysfunction by inducing apoptosis of vascular endothelial cells, as observed in SSc [36].
Studies in animal models have not demonstrated a consistent role for T cells in skin fibrosis. CD4-deficient tight-skin mice (Tsk/+) have diminished skin fibrosis suggesting that CD4+ T lymphocytes are important for the development of skin fibrosis in this model [37]. Similarly, donor T cells appear to contribute to fibrosis in the murine model of sclerodermoid GVHD [38]. Conversely, subcutaneous injection of bleomycin in severe combined immunodeficiency mice, which lack functional T cells and B cells, does not appear to affect the fibrotic response [39]. These results indicate that the role for T cells in fibrogenesis may vary between animal models and human diseases. The balance between distinct regulatory and other CD4-lineage T cells is likely important and deserves further investigation [40].
B Lymphocytes
B cell activation is of mechanistic and therapeutic interest in SSc and morphea. B lymphocytes are a component of the inflammatory infiltrate in lesional SSc skin. They are detectable upon immunohistochemical staining using CD20 surface marker, and SSc skin has elevated expression of signature genes that are consistent with the presence of B cells [41–43]. Peripheral blood B cells from SSc patients have an activated phenotype, with elevated expression of surface marker CD19 and B cell activating factor receptor (BAFFR) [44, 45]. This observation suggests that B cells in SSc may be activated by B cell activating factor (BAFF). Similar to SSc, patients with morphea also have elevated serum levels of BAFF that correlate with disease severity [45–47].
The profibrotic effects of B cell activation are likely mediated by their production of specific autoantibodies and cytokines. In SSc, B cells produce multiple autoantibodies including anti-DNA topoisomerase I, anti-centromere, anti-fibrillin-1, anti-PDGFR, anti-fibroblast, anti-endothelial cell, anti-nuclear antibodies, and others [43]. Some of these autoantibodies may have direct pathogenic roles by inducing an inflammatory response against their antigenic targets [48] (Section Scleroderma/Systemic Sclerosis). In addition, B lymphocytes produce IL-6, a cytokine that stimulates fibroblast growth and collagen production [49, 50].
Two mouse models of skin fibrosis that possess B cell abnormalities have illuminated the precise pathogenic roles of B cells in autoimmune fibrosis. Both the tight-skin mouse (Tsk/+), a genetic model of skin fibrosis, and mice subcutaneously injected with bleomycin, an injury-induced model of skin fibrosis, have activated B cell phenotypes and autoantibody production [51, 52]. Depletion of CD19+ B cells in these mouse models, as well as inhibition of BAFF in tight-skin mice, inhibit the development of skin fibrosis [52–54]. These studies suggest that inhibition of B cell activation may have therapeutic benefit for fibrotic skin diseases.
Ongoing studies in SSc patients are investigating the therapeutic benefit of B cell depletion using rituximab, a chimeric anti-CD20 antibody. Recent studies have shown a significant reduction in clinical indicators of SSc severity, including the modified Rodnan skin score, with rituximab treatment [55–57]. In addition to being potential therapeutic targets, B cell-related factors such as BAFF may also serve as biomarkers of disease progression in SSc [58].
Macrophages
Macrophages are involved in wound healing and in skin fibrosis. They are present during the inflammatory phase of wound repair [2] and in skin lesions of SSc [59, 60], acute GVHD [61], and keloidal scars [32]. Although resident macrophages are present in normal dermis, recruited monocyte-derived macrophages also contribute to wound healing [62] and SSc [63].
Macrophages are activated by profibrotic signaling factors including TGF-β, IL-13, and platelet-derived growth factor (PDGF). Functional studies in mouse models have suggested that macrophage activation is a response to factors secreted by Th2 cells, a subset of T helper cells [21]. In turn, macrophages contribute to fibrosis by secreting profibrotic cytokines including angiotensin II, CCL3, and TGF-β1 [21, 64, 65].
In mice, macrophages are present in the dermis of the bleomycin and cutaneous GVHD mouse models of fibrosis [38, 39] and are required for bleomycin-induced skin fibrosis [66] as well as for normal wound healing [62, 67]. Specifically, CCL3 produced by macrophages is critical for fibrosis in the bleomycin-induced pulmonary fibrosis mouse model [64], suggesting that this cytokine may be a key mediator of fibrogenesis in these conditions.
Signaling
Fibrosis involves altered signaling activity between all of the cellular players. In particular, inflammatory cells and fibroblasts actively secrete signaling molecules that promote the development and maintenance of pathologic fibrosis, while many of these molecules are also important in physiologic wound healing.
Signaling molecules discussed here include interleukins, chemokines, and growth factors. Recipient cells detect these secreted molecules via specific membrane-bound receptors. Generally, these signals serve to alter the target cell’s behavior (inducing migration, cell division, or another activity), identity (for example, differentiation of a mesenchymal stem cell to a fibroblast), or secretory profile (of additional signaling factors, extracellular matrix proteins, etc.). Often these changes are executed by downstream regulation of nuclear gene expression by the recipient cell. Dissecting the specific signaling pathways that result in fibrosis will provide important insight into potential targets for therapeutic intervention. As effective therapies for fibrotic diseases are sorely limited, identifying novel fibrotic signaling mediators and testing their importance in animal models is an active area of research [68].
The goal of this section is to focus on some of the prominent profibrotic signaling molecules that have been directly tested in murine models of fibrosis and wound healing. Their relevance to specific fibrosing disorders will be discussed in Section Cutaneous Fibrosing Disorders. The influence of mechanical stresses and the extracellular matrix on the signaling environment will also be discussed.
Inflammatory and Immunomodulatory Cytokines
IL-4/IL-13
IL-4 and IL-13 are cytokines produced by Th2 cells (a subset of T lymphocytes). They bind to a common receptor, IL-13Rα1 [69, 70] and both have profibrotic activity in skin.
The profibrotic effects of IL-4 include stimulating collagen production by fibroblasts, inducing expression of TGF-β, and enhancing mononuclear cell infiltration [43]. Similarly, IL-13 stimulates activation of TGF-β by cleavage from its latent binding complex, induces expression of TGF-β by macrophages, and induces expression of collagen and fibronectin by human dermal fibroblasts [21, 43, 71]. IL-13 can also induce expression of profibrotic CC-chemokines including CCL3 and CCL2 [21, 72].
Both IL-4 and IL-13 show elevated expression in lesional skin of SSc patients [71, 73], and IL-4 is expressed during normal wound healing [74]. In addition, a genetic signature consistent with IL-13 activation has been identified in the murine sclerodermatous GVHD model and in a subset of SSc patients with inflammatory disease. In these patients, IL-13 activation correlates with the severity of skin fibrosis [72]. Inhibition of IL-4 prevents dermal fibrosis in the tight-skin mouse model of SSc, and results in delayed wound healing in mice [35, 74]. Similarly, deficiency of IL-13 or its receptor IL-4Rα1 is protective against fibrosis in the sclerodermatous GVHD murine model [72].
IL-17
IL-17 is an inflammatory cytokine characteristically produced by Th17 cells, a subset of T lymphocytes. The profibrotic effects of IL-17 in skin are largely indirect. IL-17 stimulates expression of inflammatory cytokines and adhesion factors by other cell types [70]. Specifically relevant to fibrosis, IL-17 stimulates fibroblast proliferation and endothelial cell production of IL-1 and adhesion molecules. Expression of IL-17 is increased in T cells in SSc peripheral blood and skin lesions [75]. Conversely, mice lacking IL-17A have a delayed wound closure phenotype [76]. In mouse models of bleomycin-induced skin and lung fibrosis, IL-17 and other Th17 cytokines showed elevated expression, suggesting a role for Th17 mediation of the early fibrotic response [77].
IL-1
IL-1 comprises a family of inflammatory cytokines with an established profibrotic role in fibrosis of the kidney and other organs. In renal fibrosis, IL-1β, via TGF-β1, regulates epithelial-mesenchymal transition and myofibroblast conversion from fibroblasts [78]. Its role in skin fibrosis and wound healing has not been as extensively studied as in fibrosis of other organs. Both IL-1α and IL-1β are upregulated during the inflammatory phase of wound healing [79], predominantly by polymorphonuclear leukocytes and macrophages [80]. IL-1β, possibly secreted by fibroblasts, is increased in SSc. It is thought to mediate inflammation in SSc and may drive myofibroblast conversion and collagen production [81]. There is also a potential role for IL-1α from keratinocytes in stimulating fibroblast activation and contractility in fibrosis and wound healing [82]. Functional studies in animal models are needed in order to more precisely determine the sources and roles of IL-1α and IL-1β in fibrosis in vivo.
Chemokines
Chemokines (chemotactic cytokines) include CXC- and CC- molecules. These are secreted factors that bind to specific cell surface G-protein coupled receptors (GPCRs). Their potential roles in fibrosis and wound healing include inflammatory cell recruitment to the wound site, re-epithelialization, tissue remodeling, and angiogenesis [83]. Here, we will focus on two CC-type chemokines, CCL2 and CCL3. Both CCL2 and CCL3 have established profibrotic effects in murine studies of pulmonary fibrosis, possibly mediated by induction of IL-13 [21].
CCL2
CCL2 (formerly known as MCP1) acts upon monocytes/macrophages, T cells, and mast cells [83]. It is a monocyte chemoattractant and may mediate profibrotic behaviors of T cells. During wound healing, CCL2 is expressed in monocytes, macrophages, and keratinocytes [83–85]. It is also elevated in the serum in early-stage SSc, perhaps corresponding with the inflammatory phase of the disease, and is elevated in an inflammatory subset of SSc patients [72, 86]. CCL2 stimulates fibroblast expression of genes encoding TGF-β1 and type I collagen, which may be an indirect effect mediated by IL-4 produced by T cells [87, 88].
CCL2 knockout mice have delayed wound re-epithelialization and reduced angiogenesis and collagen production, without altered macrophage recruitment to the wound bed [89]. Therefore, the role of CCL2 during wound healing may be predominated by its direct or indirect effects on fibroblasts, endothelial cells, and keratinocytes, rather than by monocyte chemoattraction. CCL2 knockout mice are also resistant to bleomycin-induced skin fibrosis [66]. Furthermore, treatment with an anti-CCL2 antibody prevents the development of fibrosis in the sclerodermatous GVHD mouse [72].
CCL3
CCL3 (formerly known as MIP1α) is expressed early during wound healing in mice, corresponding with macrophage infiltration [83]. CCL3 is present at elevated levels in SSc patient serum; however, unlike CCL2, the amount of CCL3 does not correspond to the degree of skin fibrosis [90]. CCL3 knockout mice have normal wound healing [89], and CCL3 mRNA is not detected in acute adult human wounds [85]. Although this chemokine may not have a compelling role in wound healing in mice or humans, its elevated expression in SSc tissues and the possible profibrotic role of its receptor CCR5 [91] warrants its further investigation as a profibrotic mediator in human disease.
Growth Factors/Morphogens
TGF-β (Transforming Growth Factor-β)
TGF-β is the prototypic profibrotic signaling factor, and has been extensively studied in human disease conditions and animal models (Fig. 32.2). The TGF-β ligand family comprises three isoforms; TGF-β1 is the most abundant isoform in tissues. The ligand is secreted in a complex with a latent binding protein, which must be proteolytically cleaved to yield active extracellular ligand [92]. The active ligand binds to cell surface receptors, which are heterotrimers of TGF-β receptor I and TGF-β receptor II (TGFBRI and TGFBRII), stimulating their serine-threonine kinase activity [83]. Downstream intracellular transducers of TGF-β signaling activity include the canonical Smad2/Smad3-mediated response as well as non-canonical pathways [93].
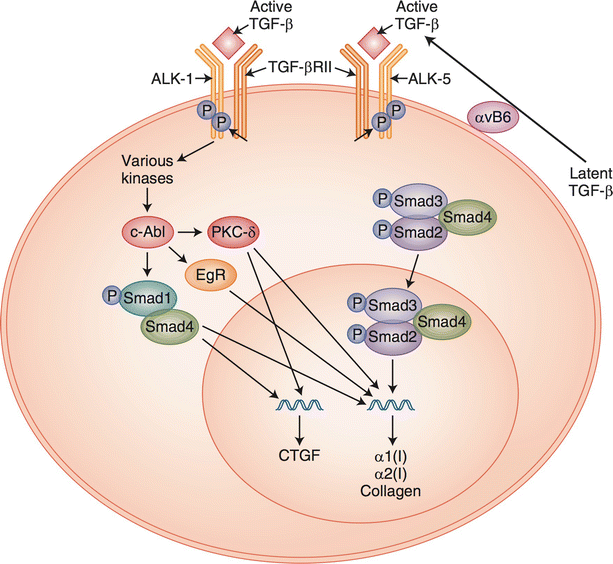
Fig. 32.2
TGF-β signaling in fibroblasts. Extracellular latent TGF-β is activated by αvβ6 integrin to its active form. Active TGF-β can signal through the classical pathway utilizing ALK-5 to phosphorylate Smad2 and -3, which then complex with Smad4. The complex is transported to the nucleus where, through additional factor interactions, critical genes can be transcribed. Non-classical signaling through ALK-1 can effect similar results through alternate pathways
The TGF-β ligand is expressed and secreted by platelets [83], activated T lymphocytes [94], monocytes and macrophages, fibroblasts, neutrophils, and epithelial cells [92]. Cells that respond to TGF-β ligand include neutrophils, macrophages, and fibroblasts [83]. The functions of TGF-β are pleiotropic and sometimes even contradictory. Depending on the cellular context, its effects may be both pro- and anti-fibrotic [21] and pro- and anti-inflammatory [92]. In the settings of fibrosis and wound healing, the prominent functions of TGF-β include promotion of monocyte activation, chemotaxis, cytokine production [92], fibroblast proliferation, expression of ECM proteins and integrins, and myofibroblast conversion [83, 95].
Elevated expression of TGF-β ligand and/or its receptors is broadly evident in fibrotic tissues including SSc lesional skin, hypertrophic scars, and keloidal scar fibroblasts [96–98]. In SSc primary fibroblasts, a TGF-β-responsive gene signature is correlated with more severe disease, providing a rationale for this signaling pathway as a therapeutic target [99]. In a transgenic mouse model, expression of constitutively active TGF-β type I receptor in fibroblasts is sufficient to cause dermal fibrosis [100]. Conversely, inhibition of TGF-β signaling results in reduced matrix deposition and scar formation in a rat model of wound healing [101] and prevents skin and lung fibrosis in the mouse model of sclerodermatous GVHD [102]. Deletion of TGFBR2 expression in fibroblasts of transgenic mice results in altered wound healing, including reduced collagen deposition with increased macrophage infiltration, and accelerated re-epithelialization and wound closure [103]. This complex phenotype highlights the importance of TGF-β signaling in regulating a variety of cellular behaviors in vivo.
Due to its many profibrotic effects on both fibroblasts and macrophages, TGF-β signaling has been under intense investigation for the treatment of fibrosis in skin and other organs. Nevertheless, neutralizing antibodies against TGF-β have not been successful in clinical trials [93]. Alternative strategies for inhibiting the profibrotic effects of TGF-β include precisely manipulating other mediators of this pathway in order to avoid off-target effects of ligand inhibition [104] and combination therapies involving inhibition of both TGF-β and other profibrotic signaling pathways [105]. A currently promising strategy involves blockade of αV integrin-mediated activation of TGF-β. This membrane-associated integrin liberates the TGF-β ligand from its inactive complex with latent binding protein (Fig. 32.2). The newly activated TGF-β then mediates myofibroblast conversion and COL1A2 expression in SSc fibroblasts [106]. Integrin blockade is anti-fibrotic in pulmonary fibrosis [107], and genetic deletion of the αV subunit in mice is protective against lung, liver, and renal fibrosis [108]. Therefore, integrin blockade and other novel manipulations of TGF-β ligand activity are of continued interest for the treatment of skin fibrosis.
Embryonic Developmental Pathways: Wnt, Notch, and Hedgehog Signaling
Classic embryonic developmental pathways are often less utilized in healthy adult tissues, but are re-activated in the contexts of tissue regeneration, wound healing, and fibrosis [19, 109]. Notch, Hedgehog, and canonical Wnt signaling are three pathways that have important gene-regulatory roles in skin development, wound healing, and fibrosis [105, 110].
The canonical Wnt/β-catenin signaling pathway is active in the presence of extracellular Wnt ligands. Activation of cell surface receptors by ligand binding results in signal transduction by intracellular β-catenin and regulates target gene expression by the Tcf/Lef family transcription factors. Wnt signaling is important for dermal reconstitution during wound healing and contributing to fibroblast proliferation, migration, motility, and matrix production [111]. Wnt signaling is activated in hypertrophic scar and keloidal scar fibroblasts and the lesional dermis of SSc and morphea patients [112–114], perhaps due to elevated expression of Wnt ligands in the skin [115, 116]. Importantly, inhibition of Wnt signaling is therapeutic in mouse models of skin fibrosis [117].
Notch signaling depends upon the interaction between transmembrane Delta-like/Jagged ligand on one cell and transmembrane Notch receptor on an adjacent cell. Mediation of this interaction by the intracellular domain of the Notch receptor results in regulation of target gene expression by the recipient cell [110]. The Notch-1 receptor is required for normal wound healing in mice, and appears to promote macrophage recruitment as well as migration of fibroblasts and vascular endothelial cells [118, 119]. This signaling pathway has elevated activity in SSc skin, and inhibition of Notch signaling is therapeutic in mouse models of skin fibrosis [120].
Hedgehog signaling involves binding of an extracellular ligand such as Sonic hedgehog (Shh) to its transmembrane receptor Patched (PTCH). This signal is transduced intracellularly to regulate target gene expression [110]. In wound healing, Shh signaling is required for normal wound closure, granulation tissue formation, and vasculogenesis [121]. This signaling pathway has elevated activity in SSc skin, and inhibition of its transducer Smoothened (Smo) is therapeutic in mouse models of skin fibrosis [122].
CTGF
Connective tissue growth factor (CTGF/CCN2) is a matricellular protein that is typically secreted by mesenchymal cells, including fibroblasts and endothelial cells [123]. Its expression is induced by TGF-β signaling (Fig. 32.2) as well as other profibrotic signaling pathways [123]. In the extracellular space, CTGF can bind to integrins as well as other secreted proteins, including TGF-β1 [124]. Based upon these properties, it follows that CTGF can mediate fibroblast and endothelial cell adhesion and potentially TGF-β1 ligand bioavailability [124, 125]. Consistent with this, CTGF is sufficient to cause increased collagen and fibronectin synthesis in human fibroblasts in vitro [126].
CTGF expression is elevated in lesional fibroblasts and plasma from SSc patients. CTGF levels correlate with the severity of fibrosis, suggesting its possible utility as a biomarker of fibrotic disease [123, 124, 126]. The role of CTGF in fibrosis and wound healing has been tested using multiple animal models. In murine models of skin fibrosis, CTGF over-expression in fibroblasts is sufficient to cause dermal, renal, and pulmonary fibrosis; and deletion of CTGF in fibroblasts protects against bleomycin-induced dermal fibrosis [127, 128]. CTGF expression is also elevated in healing wounds in rats during granulation and vasculogenesis [129]. Finally, inhibition of CTGF mRNA in a rabbit model of scar formation resulted in reduced hypertrophic scarring with lower numbers of myofibroblasts, but with maintenance of normal wound healing with respect to inflammation and angiogenesis [130].
PDGF
Platelet-derived growth factors (PDGFs) are secreted growth factors that bind to cell surface receptors that are homo- or heterodimers of PDGFRα and PDGFRβ. The PDGFRs are receptor tyrosine kinases that, when activated, trigger downstream signaling cascades including the Ras/MAP kinase (ERK) pathway. PDGF ligands are produced by fibroblasts, endothelial cells, macrophages, and platelets [131]. Fibroblasts express the PDGF receptor and respond to this signal with increased proliferation, migration, expression of extracellular matrix genes (including collagen-encoding genes), and conversion to a myofibroblast phenotype [132, 133]. Murine dermal fibrosis is associated with activation of PDGF receptors, presumably due to increased production or availability of PDGF ligands [134]. In SSc, activation of PDGF receptors can occur in response to stimulatory autoantibodies against the receptor. These autoantibodies are able to stimulate tyrosine kinase activity, type I collagen gene expression, and myofibroblast conversion in human fibroblasts in vitro [135] and therefore may be an important factor in the pathogenesis of SSc (see Section Scleroderma/Systemic Sclerosis).
A study in mice found that transgenic expression of activated PDGFRA is sufficient to cause fibrosis in skin and other organs [136]. Many more studies have evaluated readily available tyrosine kinase inhibitors (TKIs), which block PDGF signaling and other related pathways, to test for a possible therapeutic benefit of PDGF inhibition [137]. Imatinib mesylate, a TKI, inhibits PDGF signaling and reduces fibroblast proliferation, migration, collagen gene expression, and myofibroblast conversion in mouse wound healing and fibrosis models [132, 138]. One caveat is that some of the in vivo effects of imatinib and other TKIs may be due to inhibition of c-Abl (Fig. 32.2) or other protein tyrosine kinases [139, 140]. The therapeutic effects of imatinib against pre-existing dermal fibrosis in mouse models [141] provided a strong rationale for studies of its efficacy in SSc skin fibrosis. However, a phase II clinical trial did not demonstrate clinical efficacy over 6 months of treatment [142]. Additional studies addressing the profibrotic functions of specific tyrosine kinase receptors and the specificities of particular TKIs against these receptors will yield additional therapeutic options in the future.
Extracellular Matrix, Adhesion, and Mechanical Stresses
Recently, there has been a growing appreciation of the importance of tissue stiffness on cell behavior and gene expression, especially in the context of fibrosis and wound healing [143]. The fibrotic extracellular matrix has different mechanical properties from normal tissue, and this results in altered forces on fibroblasts and other cells. Elevated matrix stiffness in vitro results in increased conversion of dermal fibroblasts to a myofibroblast phenotype, and elevated expression of fibrosis-related genes such as COL1A1 and CTGF [144]. The mechanoresponsiveness of fibroblasts may be via signaling pathways that have already been established to be profibrotic, such as TGF-β/Smad2/3 (Fig. 32.2) and Wnt/β-catenin pathways [145].
Adhesion molecules, namely integrins, also mediate effects of the extracellular environment on fibroblast biology. Focal adhesion kinase (FAK), the downstream signaling transducer of integrins, is required in dermal fibroblasts for mechanosensation during scar formation; mice lacking FAK in dermal fibroblasts have reduced CCL2-mediated recruitment of inflammatory cells after wounding. This results in reduced fibrosis in a hypertrophic scar model [146]. In addition, FAK is required for dermal fibroblast expression of fibrosis-related genes including ACTA2, CTGF, and COL1A1 [147]. These findings suggest that modulating the extracellular matrix and mechanical environment during scar formation may be of therapeutic benefit. They also illustrate how physical factors can be integrated with molecular and cellular events during wound healing.
Genetic Mutations and Epigenetic Factors
Many of the classic profibrotic signaling factors exert their effects by altering expression of particular target genes. Consequently, cells and tissues in fibrosing diseases and related animal models have characteristic gene signatures that have been defined by microarray-based studies. These gene signatures are useful for identifying up- or downregulated genes that encode novel mediators of fibrosis, both in human disease [99, 148] and animal models of fibrosis [149]. In SSc, these genetic profiles can be used to divide patients into stable subtypes that predict the clinical response to treatment [150–152]. Gene expression studies can also yield insight into the pathogenesis of fibrotic disease, for example by the discovery of a B lymphocyte expression signature in SSc patient skin [42]. Next-generation sequencing technology has recently enabled the discovery of elevated fungal (Rhodotorula) gene transcripts in SSc patient skin, suggesting that this pathogen may play a provocative role in susceptibility to or in the pathogenesis of SSc [153].
In addition to altered regulation of protein-coding genes, fibrotic conditions can be associated with genetic mutations. Namely, a mutation in the protein-coding region of FBN1, which encodes fibrillin-1, causes stiff-skin syndrome [154]. This mutation is an in-frame duplication, in contrast to the single point mutation in FBN1 that causes Marfan syndrome. In addition, several single-nucleotide polymorphisms (SNPs) were found to be associated with fibrotic disease, including a SNP in the promoter region of IL1A in SSc patients [155, 156].
The presence of broad changes in gene expression, as well as the stability of the fibrosis phenotype in lesional fibroblasts cultured in vitro, has provided strong evidence for the role of epigenetic changes in fibrosis [157]. Epigenetic regulation of gene expression is often repressive in nature and can be mediated by micro-RNAs (miRNAs), methylation of DNA, and chemical modifications of DNA-associated histone complexes [158]. Down-regulation of micro-RNA (miR)-29 has been found in the skin of SSc patients and the bleomycin-induced mouse model of skin fibrosis, and miR-196a is downregulated in keloidal fibroblasts. Both of these miRs appear to inhibit expression of type I and type III collagen genes in dermal fibroblasts; therefore, decreased expression of each miR could enable elevated expression of collagen in the dermis [159, 160].
A role for DNA methylation in fibrosis has been found in dermal fibroblasts from SSc patients, where the promoter region of FLI1, which encodes a repressor of COL1A1, is heavily methylated. This methylation results in repressed expression of FLI1, which enables increased expression of type I collagen [161]. A role for decreased histone modifications in fibrosis has been identified in mice, where inhibition of methylation of lysine 27 on histone H3 (H3K27me3) is sufficient to cause dermal fibrosis, possibly by enabling expression of the profibrotic transcription factor Fra2 [162].
The increasing feasibility of genomic studies in human patients and mouse models will certainly yield additional novel pro-fibrotic mediators that may serve as therapeutic targets or biomarkers of disease progression, in addition to providing insight into how epigenetic factors might contribute to the pathogenesis of fibrosis.
Cutaneous Wound Healing
Wound healing is a physiologic process that involves many of the same cellular players and molecular pathways that promote pathologic fibrosis. Cutaneous wound healing serves to prevent infection and to repair the wound, thus preventing dehydration and restoring the protective characteristics of intact skin. In adult human skin, this process is not completely restorative, since the resulting scar tissue is mechanically weaker than native dermis and lacks functional structures, such as hair follicles and sweat glands [163]. Consequently, several studies of wound healing in humans and animal models have focused on elucidating the factors that drive fibrotic repair as opposed to faithful regeneration of skin to its original form and function.
Principles of Wound Healing
In adult mammalian tissue, partial- and full-thickness cutaneous wounds are resolved in a series of phases. Each phase involves particular cellular constituents and signaling pathways. The initial injury results in vascular disruption at the wound site, with activation of the clotting cascade producing a fibrin clot (Fig. 32.3). In the inflammatory phase of wound healing, which begins within hours of the injury, platelets in the clot release chemotactic factors that recruit neutrophils and monocytes to the site [2]. Neutrophils predominate during the early inflammatory phase to protect against infection [164]. Monocyte-derived tissue macrophages predominate later in the inflammatory phase (within days following the wound) and release growth factors that are important for new tissue formation [62].
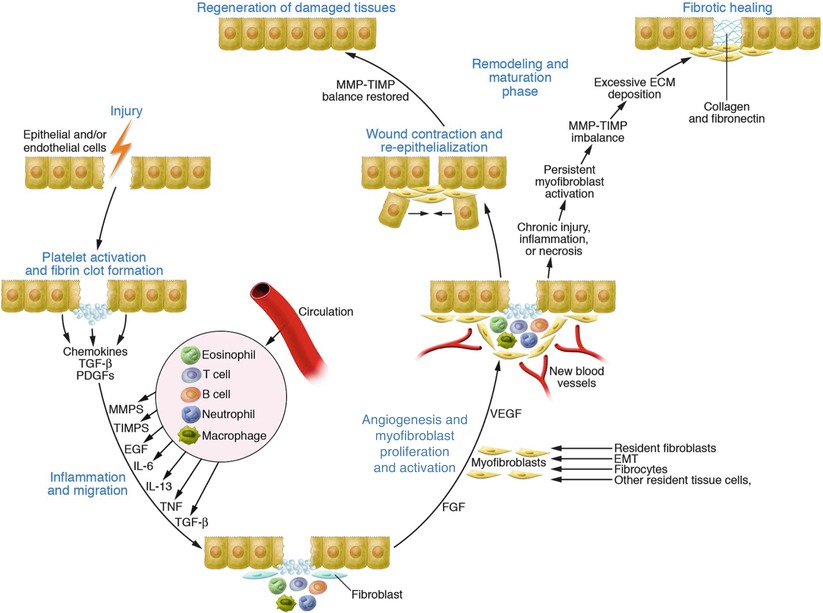
Fig. 32.3
Wound healing: regeneration and fibrosis. Following injury, signaling molecules influence the migration of fibroblasts, macrophages, and other inflammatory cells to the wound. Myofibroblasts, derived from a variety of potential sources, initiate the deposition of extracellular matrix elements and contract the wound. Provided re-injury and/or chronic inflammation do not ensue, tissue regeneration should occur. Persistent inflammation and injury signals may result in excessive matrix deposition, leading to a fibrotic scar. Many fibrosing conditions, in lieu of an actual wounding event, can elaborate similar inflammatory signals that also result in fibrosis and potential organ dysfunction (Figure utilized with permission from Wynn [68])
The new tissue that forms at the wound site during the days and weeks following the initial injury is called granulation tissue. Cells in the local inflammatory infiltrate produce growth factors that promote fibroproliferation and migration of fibroblasts, including myofibroblasts, into the wound bed [5, 6]. Angiogenesis and vasculogenesis, which involve proliferation and migration of endothelial cells, also occur during this phase. Wound repair also requires re-epithelialization over the granulation tissue and is dependent upon growth and chemotactic factors released by activated keratinocytes and underlying fibroblasts [2]. Particular populations of epidermal stem cells, which have been carefully defined in murine studies (see Section Principles of Wound Healing), contribute to the newly forming epidermis.
In the weeks to months following injury, the granulation tissue is replaced by mature scar tissue. Inflammation resolves and myofibroblasts undergo apoptosis [6]. Mature scar tissue is more collagenous than granulation tissue, better approximating the dermal extracellular matrix [2]. However, mechanical and functional properties of the skin are not wholly restored to their original state [165].
Repair and Regeneration in Animal Models of Wound Healing
Studies of wound healing in transgenic mice have defined specific populations of cells that contribute to tissue repair and regeneration. Extensive lineage-tracing experiments have labeled epidermal stem cell and progenitor populations and followed their contribution to re-epithelialization following wounding [166]. This work has shown that the healed epithelium is derived from epidermal stem/progenitor populations peripheral to the wound [167, 168].
Similar lineage-tracing studies have recently defined the mesenchymal populations that contribute to the dermal component of wound healing. PDGFRα-expressing fibroblasts residing in the upper (papillary) and lower (reticular) dermis adjacent to the wound bed contribute substantially to the healing dermis, with earlier and more robust invasion by the deeper dermal fibroblasts [169]. These fibroblasts may be recruited or supported by intradermal (subcutaneous) adipocytes, which are required for normal wound healing [170].
Mammalian fetal skin and lower vertebrates, such as zebrafish and amphibians, are champions of scarless wound healing and perfect tissue regeneration in the absence of fibrotic repair [110, 163]. One of the key differences between scarring and scar-free wound healing in these studies is that animals that can heal wounds without scars are relatively immunodeficient, suggesting that inflammation is directly related to fibrotic wound healing. Determining the specific cell types and molecular pathways engaged in tissue regeneration may point toward therapeutic strategies to help regenerate functional skin (or other organs) after injury without scarring and loss of organ function. Other than fetal wound healing, there are few examples of true regeneration in mammalian tissues. However, one such example is adult murine wound-induced hair neogenesis, in which a subpopulation of γδ T cells has been shown to be important for the formation of neogenic hair follicles in healing full-thickness wounds by regulating dermal fibroblasts to acquire more inductive characteristics [171, 172]. Continued studies of this and similar regenerative phenomena may provide provocative keys to promoting regeneration instead of scar formation during cutaneous wound healing and fibrosing skin disorders.
Cutaneous Fibrosing Disorders
Cutaneous fibrosis is a final common pathway for many disorders characterized by chronic inflammation and/or tissue injury. As noted in the preceding sections, a complex interplay of cells, growth factors, chemokines, cytokines and signaling pathways are involved in the processes of wound healing, injury recovery, and matrix homeostasis. Imbalance of any of these factors has the potential to create self-sustaining feedback loops of signaling that eventuate in fibroblast/myofibroblast activation and excess tissue matrix deposition. While a detailed mechanistic discussion of all such conditions (Table 32.1) is beyond the scope of this chapter, an in depth discussion of several common and/or well-characterized conditions is provided in the following sections to illustrate how pathological fibrosis can be triggered and sustained.
Table 32.1
Disorders commonly associated with cutaneous fibrosis
Dermatofibroma |
Diabetic digital sclerosis |
Eosinophilic fasciitis (Shulman syndrome) |
Hypertrophic scar |
Keloidal scar |
Lichen sclerosus |
Lipodermatosclerosis |
Morphea |
Nephrogenic systemic fibrosis |
Porphyria cutanea tarda (sclerodermoid) |
Radiation induced fibrosis |
Scleredema diabeticorum |
Sclerodermoid GVHD |
Scleromyxedema |
Stiff skin syndrome/congenital fascial dystrophy |
Systemic sclerosis (scleroderma) |
Toxin-associated (Spanish toxic oil syndrome, vinyl chloride) |
Scleroderma/Systemic Sclerosis
Clinical
Scleroderma (also known as systemic sclerosis, SSc) is a chronic progressive systemic disease of unknown etiology, characterized by autoimmunity, inflammation and multi-organ tissue fibrosis [173, 174]. Various lines of evidence suggest SSc is caused by interactions between the host genetic background and certain environmental factors [175]. The pathophysiology of SSc is characterized by (1) immune system activation/autoimmunity, (2) proliferative and obliterative vasculopathy, and (3) tissue fibrosis [176]. The clinical presentation of SSc is heterogeneous and can be subdivided into two entities based upon the level of skin involvement: limited (with fibrosis distal to the elbow and knee including the face), and diffuse (including proximal fibrosis) [177]. A subset of patients with limited cutaneous SSc present with “CREST” syndrome (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia). While both limited and diffuse SSc can exhibit internal organ involvement, life-threatening damage to the lungs, heart, gut, and kidneys is more common in diffuse SSc. Localized scleroderma (morphea, linear scleroderma) does not have internal organ involvement [176].
Histopathology
Skin biopsies from patients with SSc are essentially indistinguishable from those with localized scleroderma (morphea). In both entities there is thickening of collagen with loss of the clefts between the bundles. Over time, a relatively acellular hyalinized matrix replaces the fibrillar collagen. There is a variably dense superficial and mostly deep perivascular and interstitial lymphoplasmacytic infiltrate with occasional eosinophils that typically extends into the underlying subcutis by widening of the fatty septa. Loss of peri-eccrine fat is typical [178].
Autoimmunity
Oligoclonal expansion of T cells, induced by presumed environmental antigens, have been observed in SSc [179]. A variety of autoantibodies may be found in SSc [180, 181], although there is disagreement regarding their roles. Some investigators believe the autoantibodies are directly pathogenic while others suggest they may be epiphenomena, arising only after tissue damage has already occurred [182]. It is surmised that some, including antinuclear antibodies (ANA) and anti-endothelial cell antibodies, lead to the microvascular endothelial apoptosis that has been identified as the initiating pathogenic event in SSc [26, 183]. Damage to the microvascular endothelium leads to the release of proinflammatory mediators, growth factors, and chemokines.
Vasculopathy
Raynaud’s phenomenon, usually the earliest clinical sign of the disease [184], signals the presence of vasomotor dysregulation and vasculopathy. Endothelial damage triggers the activation of the coagulation and fibrinolytic cascades and ultimately the vascular occlusion, capillary necrosis, and capillary dropout seen in SSc [175, 185]. Loss of capillaries leads to tissue hypoxia, which, in turn, induces pro-angiogenic signals [186]. Other investigators have shown that exposure of endothelial cells to IFN-γ stimulates the profibrotic mediators ET-1, CTGF and TGF-β2, and that these induce the transition of endothelial cells to myofibroblasts [187].
Fibrosis and Matrix Alterations
The inappropriate leukocyte accumulation triggered by antibody-driven vascular damage leads to the elaboration of TNF (tumor necrosis factor), the release of degradative matrix metalloproteinases (MMPs), and the production of reactive oxygen species (ROS), which perpetuate additional tissue injury. With tissue hypoxia as a driving force, multiple genes for chemokines, cytokines, growth factors and their receptors are upregulated [175]. Fibroblasts respond to hypoxia by producing proteins involved in the remodeling of the ECM, such as TGF-β, thrombospondin, and CTGF [188] (see Section Growth Factors/Morphogens). In SSc, serum levels of tissue inhibitors of metalloproteinases (TIMPs) are significantly elevated, leading to an imbalance in the turnover of the ECM that favors matrix deposition [182]. In skin biopsies from SSc patients, TIMP-1 is markedly elevated while MMP-1 is not detectable [189].
The net result of these processes is the production of “damage-associated membrane proteins” (DAMPs). DAMPs, which result from cell damage, oxidative stress, and ECM remodeling, and are recognized by Toll-like receptors (TL4) on the surface of fibroblasts. One particular DAMP, a variant of fibronectin (FnEDA), seems to fulfill the functions of both structural ECM scaffold and signaling molecule [173]. FnEDA is present in significantly elevated quantities in the serum and tissue of patients with SSc. Increased FnEDA increases collagen cross-linking, promotes myofibroblast differentiation, and increases collagen gene expression. FnEDA expression, normally absent in adult tissue except during tissue injury, can also be expressed by normal fibroblasts when stimulated by TGF-β. In turn, activated fibroblasts can produce additional FnEDA, which then perpetuates a self-sustaining feedback loop that promotes the ongoing profibrotic milieu and the differentiation of a steady supply of matrix producing myofibroblasts [173].
Myofibroblasts are the primary effector cells in SSc [174], although their precise origin is debated (see Section Activated Fibroblasts and Myofibroblasts). Myofibroblasts may potentially derive from several sources (Fig. 32.1) including circulating fibrocytes (CFs), resident fibroblasts, pericytes, and epithelial or endothelial cells (via epithelial- and/or endothelial- to mesenchymal transition) [182].
The expression of genes in SSc has offered a few clues. In one gene microarray study comparing cultured fibroblasts from patients with diffuse SSc to those from normal control patients, only down-regulation of the gene MMP9 was significant after quantitative PCR analysis [190]. Another study comparing lesional and non-lesional fibroblasts in patients with diffuse SSc by gene microarray analysis showed that 26 markers performed well as predictors of fibrosis [191]. The findings of several such studies suggest that SSc is characterized by a proinflammatory gene profile that includes TGF-β and Wnt expression [192]. Activation of the Wnt pathway also stimulated an increase in fibroblast migration and proliferation, activities known to be critical in SSc [113] (Section Growth Factors/Morphogens).
Patients with SSc have also been shown to have elevation of sonic hedgehog protein (Shh) in skin fibroblasts, endothelial cells, and keratinocytes. Shh can induce myofibroblast differentiation, and its inhibition has resulted in anti-fibrotic effects in a model of SSc [122] (Section Growth Factors/Morphogens).
In human SSc, cutaneous CFs (CD45+/Collagen+) are present at high levels in the dermis, and are attracted along a chemical gradient of CCR5 (which is overexpressed by SSc monocytes). CSD (Caveolin-1 scaffolding domain peptide) has been shown in a mouse model of SSc to inhibit the accumulation of CFs, CCR5, and the CCR5 ligands CCL3 and CCL4 in the dermis. These results parallel the findings in a similar study examining lung fibrosis triggered by bleomycin injury, suggesting blockade of this CCR5 axis may have general applicability in many fibrotic conditions [91].
Nephrogenic Systemic Fibrosis
Clinical
Nephrogenic systemic fibrosis (NSF), first described in 2000, is an acquired fibrotic dermatosis associated with the use of gadolinium-containing magnetic resonance contrast agents in the setting of renal insufficiency [193]. While there is lingering controversy over the precise triggering events, evidence supports the hypothesis that certain chemical formulations of these agents are capable of promoting fibrosis in patients with transient or chronic renal disease [194, 195].
The clinical lesions of NSF typically present on the extremities (legs more commonly than arms), and occasionally involve the trunk, buttocks, and in very rare instances, the face. They consist of papules, plaques and nodules that coalesce into erythematous to brawny skin thickening, often with a peau d’orange appearance. Encompassing dermal fibrosis near joints often leads to restriction of range of motion and contractures. Pruritus and burning pain are common clinical complaints, and sometimes there is palpable warmth. It is not uncommon to observe an evolution of lesions that appears first as dependent edema, gradually becoming more erythematous, and then evolving into woody induration [193, 194].
Microscopic
Biopsies of early NSF are remarkably bland, with splaying of dermal collagen by edema and increasing numbers of banal, spindle cells in the dermis, that frequently extend into the subcutaneous tissue along fibroconnective septa [193]. There is no inflammation, although small collections of mononuclear cells may be noted adjacent to blood vessels. Fully involved biopsies show markedly thickened dermal collagen bundles associated with a complex network of CD34+ dermal spindle cells. Besides the membranous staining observed with CD34, the cytoplasm of these cells stains with procollagen I [16, 194]. Procollagen I can also be found in the dermis between these fibrocytes, along with increased amounts of dermal elastin and mucin. Collagen bundles are typically separated by clefts from one another, and a mix of thick and thin bundles is typical. These findings extend into the subcutaneous tissue along septa, and will frequently infiltrate the fascia and underlying skeletal muscle. CD68+ histiocytes are also present in variable numbers, sometimes forming multinucleated giant cells. In a small number of cases pale pink osteoid forms around protruding elastin fibers creating the so-called “lollipop” body, which is considered virtually pathognomonic for NSF. On occasion these bodies will mineralize [194]. Rarely, widespread mineralization of dermal collagen can occur. Initial tissue staining studies showed markedly increased TGF-β1 mRNA by in situ hybridization throughout the dermis of all patients examined [196].
Triggers
Epidemiological evidence has convincingly established that NSF is a disease of the renally impaired. Patients at risk for NSF have acute kidney injury (AKI) or an estimated glomerular filtration rate (eGFR) of less 30 ml/min/1.73 m2. Investigators have concluded that NSF is triggered by the administration of gadolinium based contrast agents (GBCAs) to patients with the requisite degree of renal impairment. Gadolinium (Gd) is a paramagnetic element of the lanthanide series, making it an ideal contrast agent in magnetic resonance imaging (MRI) studies. As ionized Gd3+ is considered toxic because of its potent calcium channel blocking abilities, the ion is chelated with a proprietary organic ligand that may be linear or macrocyclic in structure. This reduces the toxicity of Gd while simultaneously facilitating the excretion of the chelated complex, which in a patient with normal renal function has a half-life of about 90 min. Over the years since the discovery of NSF, there have been several brands of GBCA available on the market in the United States and abroad. Epidemiological evidence has associated the vast majority of cases of NSF with three brands of GBCA, these being agents with a linear organic ligand. The use of these agents in the renally-impaired has been specifically contraindicated by regulatory agencies, leading to a very rapid decline in new cases of NSF since 2010 [195].
While these agents were still being used in the at-risk population, it became clear that only 2–6 % of those at risk actually contracted NSF upon exposure to one or more doses of the agent. The reasons for this variability in the at-risk population are unknown, although some have surmised there might be a genetic propensity or some other as yet unrecognized risk factor for NSF [197].
Cell Culture Studies
Epidemiological and clinical aspects of NSF have implicated excess extravasated GBCA as the initial triggering event in NSF. Electron microscopy with energy dispersive spectroscopy has identified Gd within the macrophages seen in abundance in early lesions of NSF [198]. The dominant cells in the histopathological specimens from NSF, macrophages and fibrocytes, have been extensively studied by cell culture techniques.
Fibroblasts/Fibrocytes
In 2003, a compelling case was made for the involvement of bone marrow-derived circulating fibrocytes in NSF [16]. Circulating fibrocytes, defined by their dual marker positivity for CD34 and procollagen I, participate in normal wound healing and have been shown to be active in several fibrotic processes. In their fully mature form they histologically resemble and likely supplement the activities of resident dermal fibroblasts.
When added to the serum of normal cultured fibroblasts, Omniscan™, the GBCA associated with the majority of NSF cases, increases the fibroblast proliferation by more than two-fold. It also increases MMP-1 (1.5-fold) and TIMP-1 (12-fold). While individual cell procollagen production was not directly stimulated, the cumulative effects of increased fibroblast proliferation and altered collagen turnover were judged to contribute to the fibrosis of NSF [199]. While both MMP-1 and TIMP-1 are increased, the vast excess production of TIMP-1 effectively outweighs the collagenolytic activity provided by MMP-1 [200]. The increased TIMP-1 and absent MMP-1 pattern has been directly visualized in immunohistochemical studies from NSF biopsy specimens, corroborating the culture observations [189].
Dermal fibroblasts obtained from patients with NSF display a marked increase in the production of type I (2 to 3-fold) and type III (3.5 to 6-fold) collagen, fibronectin (3 to 7-fold), and hyaluronic acid (4 to 5-fold). This effect was maintained after 11 culture passages, suggesting the effect might be a stable epigenetic change. Collagen I production was increased at both the translational and transcriptional levels. The increased transcriptional activity of the COL1A1 promoter was due to increased cREL, NF1, and CBF transcription factor binding [201]. In NSF, cREL and NF1 appeared to show the most binding activity, in contrast to CBF and Sp1, which are the most active transcription factors in SSc [202]. The elevation of TGF-β combined with the markedly increased cREL component of the NF-kB family of transcription factors suggests that there may be an important interaction between these pathways in NSF [201].
A culture study conducted on normal human fibroblasts concluded that Gd3+ directly or indirectly modulates signaling through the PDGF receptor. The investigators postulated that Gd3+ attaches to an available protein or some other “carrier molecule” that allows it to remain soluble long enough to interact with the PDGF receptor. They also noted that fibrotic changes similar to those seen in NSF have been produced in rats exposed to other chelated lanthanides, suggesting that the observed effects are not Gd specific. This observation offers the intriguing possibility that metallic elements other than Gd, in the correct clinical setting, may be capable of interacting with the PDGF receptor to produce similar fibrotic outcomes [203].
Another study examining normal human fibroblasts concluded that the PDGF stimulatory effects of Omniscan™ could be abrogated by exogenously administered CCN3 (also known as nephroblastoma overexpressed gene). CCN3 is a matricellular protein that is able to suppress CTGF/CCN2, a molecule critical to the initiation and progression of renal fibrosis [204]. The authors noted that Omniscan™ suppressed the endogenous production of CTGF/CCN2, leading to a permissive environment for both PDGF and TGF-β driven fibrogenic processes [205].
Monocyte/Macrophages
In human peripheral blood monocyte studies, Gd compounds caused significant upregulation of multiple cytokines and growth factors, including VEGF (253-fold), IL-13 (68-fold), IL-4 (28-fold) and IL-6. TGF-β was increased two to three fold (depending on the agent used) and IFN-γ was elevated up to 50-fold. The timing and magnitude of the effects varied with the agents used and the subjects, although intrasubject response was stable [206]. Normal fibroblasts cultured with media conditioned by peripheral blood monocytes that had been exposed to Omniscan™ displayed a dose-dependent increase in production of type I procollagen [206].
In a gene microarray evaluation of normal human macrophages incubated with 50 mM Omniscan™, 551 differentially expressed genes were noted. There was potent stimulation of CCL8 (669 ± 108-fold), CXCL10 (401 ± 72-fold), CCL2 (245 ± 36-fold) and CXCL11 (551 ± 48-fold). Pathway analysis proved that exposure to Omniscan™ elicits chemokine gene expression in macrophages that is dependent on NFkB activation [207]. As it is widely accepted that circulating fibrocytes home to the skin following a specific chemokine gradient, the investigators concluded that macrophages stimulated by this mechanism produce the chemokines needed for fibrocyte chemotaxis to skin.
In addition, the effects of Omniscan™ and gadodiamide in macrophages are dependent on TLR4 and TLR7. It is hypothesized that these agents possess a unique, specific molecular shape or pattern that renders them capable of this interaction. Initial engagement of TLR4 by Omniscan™ at the macrophage surface could induce and increase the rates of phagocytosis, which could amplify its TLR signaling capacity through the engagement of TLR7 within the endosome [208]. As fibroblasts also express surface TLR4, it is possible that some of the direct effects of Gd compounds on fibroblasts could be due to engagement of these receptors (Fig. 32.4) [208].
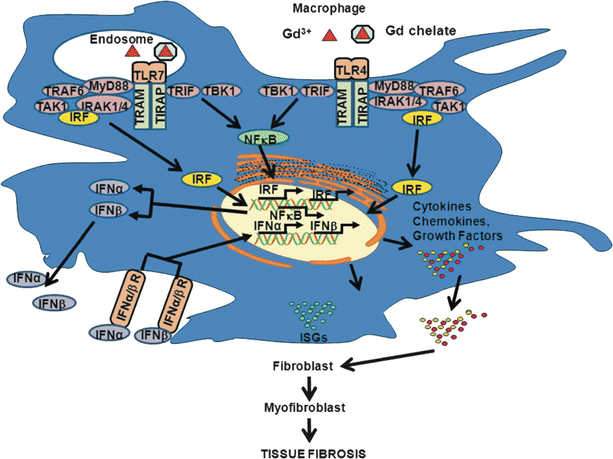
Fig. 32.4
Gadolinium based contrast agent (GBCA) induction of a proinflammatory/profibrotic phenotype in normal human macrophages. Gd3+ or GBCA is phagocytosed. Within the acidic environment of the endosome, dechelation is facilitated. Either Gd3+ or GBCA interact through TLR 4 and 7 to upregulate the NFkB protein complex, leading to the production of proinflammatory and profibrotic factors that are subsequently secreted by these “activated” macrophages. These secreted factors facilitate the development and migration of myofibroblasts from a variety of tissue sources, which then result in sustained and dysregulated matrix production (Figure utilized with permission from Wermuth and Jimenez [209])
These investigators conclude that all Gd compounds examined were capable of inducing expression of genes associated with TLR signaling and that the shape of individual GBCAs, in combination with direct effects of free Gd3+ likely both contribute to NSF pathogenesis [209].
Animal Modeling
Utilizing a rat model of NSF exposed to gadodiamide (the active ingredient in Omniscan™) investigators have induced high levels of serum cytokines, the most significantly elevated being CCL2 (6.5-fold) and CCL7 (6.8-fold), macrophage inflammatory proteins CCL4 (2.8-fold) and CXCL2 (2.5-fold), TIMP-1 (3.6-fold), TNF-α (3.4-fold), osteopontin (6.1-fold), and VEGF (1.6-fold) [210]. The role of osteopontin is particularly intriguing to these investigators as it is known as a mediator in lung disease, interacting with receptors (CXCR4, CCR7) of circulating fibrocytes [211].
These same investigators also noted a drop in serum albumin that correlated with marked vascular permeability within 6 h of gadodiamide exposure [212], a finding that parallels the early edema and erythema seen in NSF patients clinically. This tissue vascular leakage facilitates the extravasation of GBCA into the extravascular space, where it is subsequently phagocytosed by macrophages.
Fascinating work completed using mouse modeling has shown that Gd3+ and GBCA activate the NLRP3 inflammasome, primarily in undifferentiated and LPS-primed M2 macrophages [213]. The NLRP3 inflammasome is involved in the rapid recruitment of inflammatory monocytes and granulocytes to the site of Gd deposition in vivo, and can be stimulated by extracellular calcium and Gd3+ through a calcium-sensing receptor (CASR) [214]. GBCAs also induce the production of mature IL-1β by macrophages in vitro through NLRP3-dependent mechanisms. IL-1β can promote fibrosis by stimulating fibroblasts and by inducing TGF-β1 [213].
Keloids
Clinical
Keloids are defined as exuberant scar tissue formation extending beyond the area of original tissue injury [215]. Clinically, keloids are firm broad nodules with a shiny surface that often have a claw-like appearance beyond the original site of injury [215]. Keloids may show a delayed onset, sometimes forming months after the original injury. They can be painful or pruritic [215]. Persons of African or Asian heritage are reportedly ten times more susceptible to keloids than are whites [216].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


