Patients with craniofacial microsomia comprise a diverse clinical cohort that requires individualized attention and surgical consideration that benefits from multidisciplinary team management to optimize functionality and esthetics. Specific concerns regarding airway, vision, feeding, growth, hearing, speech, development, and quality of life may require intervention. The full reconstructive ladder may be utilized in the care of these patients. Ultimately, tailoring surgery to optimize final facial symmetry while minimizing the burden of surgical interventions serves these patients well.
Key points
- •
Phenotypic characteristics and severity in craniofacial microsomia vary widely but typically include involvement of the orbit, mandible, ear, facial nerve, and soft tissue.
- •
Diagnostic criteria are ill defined and several clinical classification systems exist for the facial features in craniofacial microsomia.
- •
Clinical care teams and approaches to management should be multidisciplinary, while interventions should be individualized to fit the patient’s phenotype, functional concerns, and preferences.
- •
Further research is required to evaluate optimal timing and type of treatments. Multicenter, international research teams continue to form to facilitate advances in care protocols and disease understanding.
Introduction
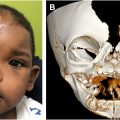
Craniofacial microsomia (CFM) is a clinical term that refers to patients with absence or underdevelopment of facial structures that arise from the first and second pharyngeal arches. The CFM spectrum includes hemifacial microsomia, oculo-auriculo-vertebral spectrum, otomandibular dysostosis, lateral facial dysplasia, and first and second branchial arch syndrome. Goldenhar syndrome is often considered to be part of the CFM continuum and also includes epibulbar dermoids and vertebral anomalies. , CFM occurs in as many as 1:3000 to 1:5000 live births and is the second most common congenital disorder of the face, behind cleft lip/palate. Prevalence is higher among boys and children from Hispanic and Native American families. While phenotypic presentations can vary widely, hypoplasia of the mandible, maxilla, ear, orbit, facial soft tissue, and/or facial nerve is often observed. Extracranial anomalies in the central nervous system (CNS), skeletal, cardiac, lung, gastrointestinal, and renal systems are also common. Functional implications include feeding and airway difficulties, altered facial movements and appearance, hearing deficits, and vision and ocular problems. Given the wide range of potential clinical needs, these patients benefit from management by multidisciplinary care teams.
This review is not meant to be an exhaustive summary of craniofacial surgical techniques that have remained largely unchanged, but instead to summarize general treatment concepts and to highlight significant developments.
Etiology and pathogenesis
There are several suspected causes of CFM, including inherited genetic abnormalities, teratogen exposure, and vascular injury in utero. During the first 6 weeks of gestation, disruption in the development of skeletal, muscular, and neural structures derived from the first and second pharyngeal arches results in the phenotypic features in CFM. The first branchial arch gives rise to the maxilla, mandible, zygoma, trigeminal nerve, muscles of mastication, and the anterior portion of the ear (tragus, root, and superior helix) as well as the malleolus and incus. The second branchial arch develops into the hyoid bone, the styloid process and facial nerve, the facial muscles and most of the ear (helix, antihelix, antitragus, and lobule), as well as the stapes. Development is contingent on continuous communication between mesenchymal cells; aberrancies in this process due to various mechanisms can thus result in hypoplasia or aplasia of the affected structures. , In 1973, Poswillo demonstrated that by inducing a hematoma of the stapedial artery (blood supply to the first and second pharyngeal arch) in mice using a teratogen (trazine) in the sixth week of gestation, he could produce the CFM phenotype. The authors concluded that a vascular insult to the developing arches could be an underlying mechanism for CFM. Furthermore, they posited that the phenotypic variation in CFM may reflect the degree of vascular disruption and its ability to regenerate. Since then, other studies have identified additional risk factors: maternal use of vasoactive medications, hormonal therapy, second-trimester smoking, diabetes mellitus, hypothyroidism, and use of reproductive technology. , , In addition, a variety of genetic causes with both autosomal dominant and recessive inheritance patterns have been described. Abnormal neural crest cell migration, proliferation, and differentiation are proving to be another leading theory of CFM pathogenesis. ,
Clinical presentation
The CFM diagnosis is based on physical examination findings of hypoplasia, aplasia, or malformation of one or more of the structures deriving from the first or second branchial arch (ie, external ear, mandible, temporal bone, zygoma, middle ear, facial musculature, facial nerve supply, and other adjacent bony and soft tissues). Ocular deformities include strabismus, anophthalmia, microphthalmia, orbital asymmetry, cleft eyelid, and exophthalmia. Auricular abnormalities include preauricular appendage, preauricular fistula, microtia, ear asymmetry, and external auditory canal atresia. It remains debated in literature whether isolated microtia is a distinct entity or minor variant of CFM. Additional deformities of the first and second pharyngeal arches include cleft lip and palate, bifid tongue, mandibular hypoplasia, maxillary hypoplasia, malocclusion, and dental malformations. Common associated functional concerns include feeding difficulties, obstructive sleep apnea, and dysphagia.
The anomalies may be unilateral or bilateral and range in severity. Bilateral presentations are seen in approximately 10% of patients and are most common with autosomal dominant inheritance patterns.
Patients with CFM often display extracranial abnormalities as well, affecting the neurologic (5%–15%), cardiac (14%–47%), genitourinary (5%–6%), pulmonary and gastrointestinal (10%), and skeletal systems (40%–60%).
The wide phenotypic variability challenges investigations on CFM etiology and treatment outcomes. Moreover, diagnostic criteria do not currently exist. This lack of consensus further impacts our ability to evaluate disease prevalence and existing treatment protocols. The following minimal diagnostic criteria have been recommended (1) ipsilateral mandibular and auricle defects and (2) asymmetrical mandibular or auricle defects in association with (1) 2 or more indirectly associated anomalies or (2) a positive family history of CFM. ,
In recent years, 2 sets of phenotypic criteria for CFM have been developed for clinical research. They were developed independently, based on consensus among distinct multidisciplinary health care providers with expertise treating and researching patients with CFM. The multicenter consortium “Facial Asymmetry Collaborative for Interdisciplinary Assessment and Learning (FACIAL),” which started in 2009, was established to develop standardized definitions and study protocols to facilitate clinical research on CFM. This collaborative created eligibility diagnostic criteria for research based on the different CFM features. A similar, newer initiative was created in 2017 by the “International Consortium for Health Outcomes Measurement (ICHOM),” which aims to implement a global standard set to obtain comparable data for benchmarking and research. A 2023 multicenter study evaluated the FACIAL and ICHOM CFM criteria with an existing database of patients with CFM to assess the sensitivity of these criteria sets and study characteristics of patients with CFM that do not reach criteria. , Authors found that both criteria show high sensitivity (FACIAL 94.4% and ICHOM 99.6%) with a fair agreement between both criteria. In this studied cohort, the ICHOM criteria tend to be most accurate. They concluded that both criteria are considered useful for future studies on CFM for outcome comparisons.
Evaluation
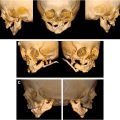
Historically, the gold standard for assessing facial asymmetry is the 2-dimensional posteroanterior cephalogram that can be used along with lateral cephalograms and panorex images. , Traditional photography can document facial appearance throughout growth and treatment. Three dimensional techniques including cone beam computed tomography, stereophotogrammetry, laser scanning, and contact digitalization can provide more detailed, precise, and accurate measurements and are often used for surgical planning and fabrication of skeletal models.
Patients with CFM present with signs and symptoms associated with their individual phenotype. Hypoplasia of the pharynx, larynx, esophagus, mandible, and mastication muscles can lead to airway obstruction, feeding difficulties, and failure to thrive. , Thus, polysomnography and swallow evaluation by a speech-language pathologist are critical, particularly in cases of micrognathia and OSA symptoms. It is important to distinguish mandibular hypoplasia of CFM from that associated with Pierre Robin sequence (PRS) given the growth potential in patients with PRS and potential success with temporizing measures in early infancy such as prone positioning, nasopharyngeal airways, and tongue–lip adhesions. Relatedly, features of CFM such as microtia and mandibular hypoplasia overlap with several other established syndromes such as coloboma, heart defect, atresia choanae, restricted growth and development, genital abnormality, and ear abnormality; Treacher–Collins; Nager acrofacial dysostosis; PRS; Parry–Romberg; and others.
Additional screenings include ocular examinations, audiograms, and perceptual speech analysis. Extracranial screening examinations, such as psychosocial assessments, cervical spine radiographs, and renal ultrasound, should also be performed. In cases with suspected genetic inheritance, chromosomal analysis and genetic counseling can also be offered.
Classification
Due to the variable presentation, several classification systems have been developed to differentiate patients and facilitate discussions of treatment, prognosis, and outcomes.
The first classification scheme was proposed by Pruzansky and included 3 severity grades focused on the mandible: grade I describes hemimandibles smaller than the normal side; grade II refers to mandibles with a grossly distorted (size/shape) but identifiable condyle, ramus, and sigmoid notch; grade 3 mandibles have grossly a distorted ramus with loss of landmarks, or agenesis. Later, Kaban and colleagues , modified this system to better describe the position of the temporomandibular joint (TMJ).In this system, grade I still indicates a small mandible. Grade II is divided with the IIA mandible displaying a short ramus, but the glenoid fossa is in a normal position, while the IIB mandible has a malpositioned glenoid fossa that is typically inferiorly, medially, and anteriorly displaced. Grade III has an absent TMJ. Kaban and colleagues felt that the IIB mandible required reconstruction of the TMJ, whereas the IIA mandible did not. This classification system, as well as others that incorporate 3 dimensional mandible images, is widely used and can be helpful guides to treatment of the mandibular deformity in CFM.
Other classification schemes were developed to encompass the additional facial features involved. In 1987, David and colleagues , proposed the SAT (skeletal malformations, auricular involvement, and soft tissue defects) system to grade the severity of these defects and provide recommendations for surgical interventions based on scores.
The most comprehensive and widely used classification system, OMENS, was developed by Vento and colleagues in 1991. It scores the severity of abnormality seen in the 5 major manifestations of CFM from 0 (normal/no obvious deficiency) to 3 (most severe): orbit, mandible, ear, nerve, soft tissue. Notably, the authors found a positive association between mandibular hypoplasia and the severity of orbital, auricular, neural, and soft tissue involvement. The OMENS+ system was developed to incorporate extracranial abnormalities. Subsequently, an OMENS pictographic representation was designed and modified to the phenotypic assessment tool, Phenotypic Assessment Tool- Craniofacial Microsomia (PAT-CFM), to improve ease of use.
Surgical treatment
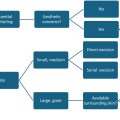
No single surgical protocol exists due to the variability of clinical needs seen in patients with CFM. Treatments targeting compromised functionality (such as airway obstruction) should take precedence over esthetic issues and may require earlier intervention. That said, restoration of facial symmetry and contouring can be a significant aesthetic challenge for these patients. The Craniofacial Center at Seattle Children’s Hospital has proposed and previously published a comprehensive timeline for the medical and surgical management of individuals with CFM. It outlines considerations for various symptoms/issues over time that can be used as a general guide for practitioners as well as patients or families as they try to understand the interventions that may be needed overtime. It is essential to understand how surgical interventions are impacted by expected facial growth patterns, timing of dental eruption, and psychosocial factors. Again, many of the specific reconstructive techniques required to address the craniofacial abnormalities will not again be detailed here, but rather we will highlight significant considerations for the common surgical procedures later.
Orbit
Assessment of the visual axis in infancy is necessary, and treatment of epibulbar dermoids may be required. Eyelid colobomas should be corrected if risk of exposure keratitis, and ultimately blindness, is a concern. Severe orbital dystopia (asymmetry in size and/or position of the orbits) is corrected via box osteotomy, typically not until the age of 6 years.
Mandible
Controversy regarding management of mandibular defects remains. Management of the TMJ and mandible in terms of timing of treatments, approaches, and specific surgical technique continue to vary between surgical centers. Mandibular distraction is sometimes offered for infants with failure to thrive related to mandible-level airway obstruction in an effort to avoid tracheostomy. Prior to this, a complete evaluation of infant growth, feeding, and airway patency is critical.
For Pruzansky type I mandibles, surgical intervention can often be postponed until skeletal maturity. Treatment can range from orthodontics alone to bimaxillary orthognathic surgery.
For Pruzansky type II mandibles, treatment needs vary based on subgroups. In IIa mandibles, vertical lengthening is needed. Mandibular distraction osteogenesis (MDO) can be used to successfully lengthen a short ramus with subsequent orthodontics for management of a maxillary cant. Distraction osteogenesis in the mandible was introduced in an attempt to address the soft tissue as well as osseous deficiencies seen in these patients. Alternatively, osteotomy and interposed bone graft and orthognathic surgery at skeletal maturity can be considered. For Pruzansky IIb mandibles, the status of the TMJ is critical. Classically, zygomatic arch and glenoid fossa reconstruction was performed with cranial bone grafts and conchal cartilage grafts, and ramus reconstruction used costochondral bone grafts. Some investigators stage reconstruction out of concern for ankylosis.
For Pruzansky type III mandibles, the goal of reconstruction is again to create adequate posterior ramus height as well as a functional TMJ. Costochondral bone grafts or vascularized free tissue transfer can provide the additional bone stock, although both methods have been associated with ankylosis. Costochondral grafts can have unpredictable growth and resorption, potentially due to the lack of regional soft tissue and decreased vascularity. This procedure is typically done when the patient begins to show an occlusal cant in the maxilla, generally corelating with dental eruption around the age of 2 to 5 years. MDO can again be used in combination with an initial bone graft to create a sufficient ramus to distract. Whether a graft or flap is used, the bone is fixated to the remnant angle or body and allowed to heal and then can be followed by distraction. We prefer to distract the native mandible rather than the graft whenever possible and use the grafted area to secure the footplates of the distraction device.
It is essential for the surgeon and orthodontist to coordinate care in these cases. Lengthening the mandible via distraction creates an open bite that must be maintained until maxillary dentition can be brought down into a stable occlusion.
Ultimately, many patients will benefit from orthognathic surgery to level occlusal cants and improve skeletal asymmetry. This should be delayed until skeletal maturity if possible in order to reduce the risk of recurrent asymmetries. Given the complex movements required in these patients, patient-specific preoperative planning is critical. Virtual surgical planning has largely replaced conventional planning techniques in most centers given its ease of simulating several different movements, detecting interferences, and clear perioperative measurements at anatomic landmarks that can be used for guidance intraoperatively.
Ear
Ear anomalies include external ear malformations (such as microtia), middle ear malformations and atresia, and branchial remnants or sinus tracts. The severity of the external ear malformation may predict the degree of middle ear involvement.
For mild hypoplasia (OMENS E1 ears), reshaping cartilage with rasping, burr scoring, and/or suture stabilization may suffice. Contralateral otoplasty can be considered for symmetry.
For E2 and E3 ears (absent EAC with conchal hypoplasia, and malpositioned lobule with absent auricle, respectively), many surgeons prefer to discard the remnant cartilage and proceed with total ear reconstruction; these options include alloplastic ear reconstruction (most commonly porous polyethylene), autologous rib graft reconstruction, and prosthetic devices with osseointegrated implants. Alloplastic reconstructions have shown favorable long-term results despite initial reports of extrusion and infection, leading to a wider use and acceptance. Importantly, these reconstructions avoid the need for a donor site and can be completed earlier and in a single stage. Simultaneous external ear and placement of a bone-anchored hearing aid is an option to be considered by the multidisciplinary team. Generally, autologous reconstruction is performed when the ear reaches adult size and the patient has adequate costal cartilage for the framework (between ages of 6 and 10 years).
Soft Tissue
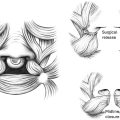
Treatment of macrostomia and branchial remnants is often performed when the patient is cleared for general anesthesia (6–12 months). Macrostomia (Tessier VII cleft) repair should address the clefted mucosa and skin as well as reconstruct the orbicularis oris sphincter with commissuroplasty to facilitate oral competence. For branchial clefts, complete excision of any cartilaginous stalk is necessary to prevent further growth after excision.
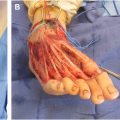
Facial nerve palsy is typically partial and unilateral but may be bilateral. The OMENS classification categorizes nerve involvement into upper, lower, and total loss. Pediatric patients can greatly benefit from dynamic reanimation procedures. Initial assessments must determine the patient’s ability to protect the cornea; medical management with lubricating drops or overnight ointments may suffice, and surgical intervention is beneficial if not. Facial reanimation is considered in cases of affected buccal and marginal mandibular branches to provide smile and mouth movement. The most common dynamic procedure performed for patients with CFM and unilateral palsy is the 2 stage facial reanimation procedure involving a cross-face nerve graft (sural nerve donor) using redundant buccal branches from the unaffected side to ultimately innervate a vascularized muscle flap (most often a gracilis free flap) to create a spontaneous smile. Disadvantages include the prolonged surgical course, involvement of the unaffected side as a donor, and weakness of the smile. Locoregional muscle flaps (masseter or temporalis) are alternatives that avoid multiple stages and can function immediately. Disadvantages include weak strength and limitations on vector of pull and non-spontaneous smile (innervated by cranial nerve V). This surgery is planned around other surgeries that these patients need; typically microtia and other major craniofacial/orthognathic surgeries are performed prior to facial reanimation.
Finally, soft tissue deficiency must be addressed as well. Alloplastic implants can be utilized to camouflage asymmetry in an off-the-shelf implant or with a patient-specific design. Alternatively, local or distant flaps (such as an adipofascial free flap), dermal fat grafts, or structural fat grafting are all reconstructive options that can be effective in these patients depending on their specific need. Free flaps are generally reserved for severe cases and revisions for debulking should be anticipated. Described donor sites include scapula, parascapular, groin, omentum, anteriolateral thigh flap (ALT), and deep inferior epigastric perforator flap (DIEP) flaps. Fat grafts often require serial treatments due to resorption over time, but offer advantages of precise augmentation, minimal donor-site morbidity, and limited scarring. These treatments can often be coordinated with other procedures throughout childhood to minimize recovery and number of procedures, as well as to improve facial symmetry during development.
Future considerations
Development of robust, evidenced-based treatment guidelines is challenging given ongoing questions regarding disease etiology, the lack of consensus on diagnostic criteria, and the lack of large population studies to inform care protocols. Previously highlighted efforts include the creation of the FACIAL, which is an interdisciplinary and multicenter research network, as well as the ICHOM working group for CFM that developed outcome metrics for function, appearance, psychosocial concerns, and treatment burden. More recently, the Craniofacial Microsomia: Accelerating Research and Education program was established to investigate psychosocial health concerns for patients with CFM and is establishing a registry to build a longitudinal database and develop an international community around CFM.
Summary
Patients with CFM comprise a diverse clinical cohort that requires individualized attention and surgical consideration that benefits from multidisciplinary team management to optimize functionality and esthetics. Specific concerns regarding airway, vision, feeding, growth, hearing, speech, psychosocial development, and quality of life may require intervention. The full reconstructive ladder may be utilized in the care of these patients. Ultimately, tailoring surgery to optimize final facial symmetry while minimizing the burden of surgical interventions serves these patients well.
Clinics care points
- •
Phenotypic characteristics and severity in CFM vary widely but typically include involvement of the orbit, mandible, ear, facial nerve, and soft tissue.
- •
Diagnostic criteria are ill-defined and several clinical classification systems exist for the facial features in CFM.
- •
Clinical care teams and approaches to management should be multidisciplinary, while interventions should be individualized to fit the patient’s phenotype, functional concerns, and preferences.
- •
Further research is required to evaluate optimal timing and type of treatments. Multicenter, international research teams continue to form to facilitate advances in care protocols and disease understanding.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


