Fig. 23.1
Contact allergens and irritants cause tissue stress and damage. ROS are induced and stressed or damaged cells release and or/produce danger signals such as PRR-activating DAMPs. Additional danger signals are derived from the extracellular matrix. Consequently skin inflammation results and DCs are activated and migrate from the epidermis to the dermis and then to skin draining lymph nodes. Due to the chemical modification of proteins contact allergens form T cell epitopes that are presented on activated DCs in the lymph node and prime contact allergen-specific T cells. This concludes the sensitization phase of ACD. The recruitment of activated effector/memory T cells to the skin upon repeated contact with the contact allergen leads to eczema. Irritants cause eczema without induction of an allergen-specific T cell response
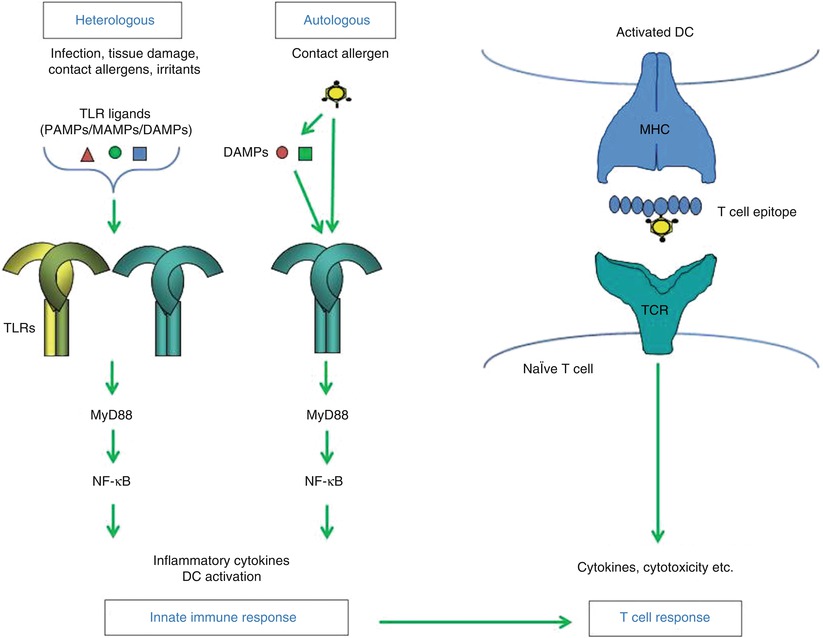
Fig. 23.2
Heterologous innate immune stimulation. Contact allergens can directly or indirectly provide autologous innate immune stimulation for example by triggering TLRs leading to activation of NF-kB and production of inflammatory cytokines and DC activation and migration. Heterologus innate immune stimulation can be provided for example by infection (e.g., PAMPs), tissue damage (DAMPs), irritants and other contact allergens that trigger the same signaling pathways via the same or different TLRs. As a result autologous innate immune stimulation can be amplified or – when absent – be substituted by heterologous innate immune stimulation. Due to T cell epitope formation by the contact allergen a T cell response is induced by activated DC when the innate immune stimulation is sufficiently strong. Upon repeated allergen contact contact allergen-specific effector/memory T cells enter the skin and exert effector function such as cytokine production and cytotoxicity
Diagnosis of Allergic Contact Dermatitis
The diagnosis of ACD is ideally based on the patient’s history, the clinical presentation and a positive patch test reaction to the suspected contact allergen. In every day practice this ideal situation is rarely present. The clinical presentation, the affected areas and the distribution may be suggestive of ACD, while morphological skin changes and histology do not allow a definite differentiation between ACD and other forms of dermatitis, such as ICD, atopic dermatitis or seborrheic dermatitis. For the identification of relevant contact allergens a detailed patient history is of utmost importance. This must include detailed information on the work environment, recreational activities, medication, use of cosmetics, emollients, detergents and other substances that the patient has been exposed to prior to the onset of the dermatitis.
The mainstay for the diagnosis of ACD is the patch test in which under standardized conditions contact allergens are applied to the healthy skin of the patient with the goal to reproduce the allergic eczematous skin reaction. Prerequisite for patch testing with suspected contact allergens is that skin reactions during acute or chronic ACD have been sufficiently controlled or even better resolved by topical or systemic anti-inflammatory treatment. Patch testing during ongoing ACD produces increased numbers of false positive patch test reactions and thus should be avoided.
The standard procedure of the patch test involves application of the contact allergen in the corresponding vehicle (vaseline for lipophilic, aqueous solution for hydrophilic allergens) in a Finn chamber to the skin (usually skin of the back) of the patient for 24 h or 48 h. The first reading is taken at 48 h, a second reading is recommended at 72–96 h after initiation of the test. Standardized criteria for reading the patch test reactions have been developed by the DKG and allow a grading and interpretation of the reaction (Table 23.1). Like all test systems the patch test has also a number of pitfalls and limitations and may generate false positive or false negative results. False positive results may occur in patients in whom the ACD has not sufficiently been treated or in patients that display a very strong sensitization. In these situations multiple chemically non-related allergens can induce false positive reactions. This phenomenon is described as “angry back” or excited skin syndrome and most likely reflects the fact that the skin displays reduced thresholds to the irritative capacity of these unrelated allergens.
Table 23.1
Grading of patch test reactions according to the German Working Group for Contact Dermatitis (DKG)
Grading | Morphology | Interpretation |
|---|---|---|
− | No changes | Negative |
ir | Sharply demarcated erythema, blister, erosion, necrosis, ulcer | Irritative |
? | Only erythema (allergic or irritative) | Questionable |
F | Follicular papules, and/or pustules | Questionable |
+ | Erythema, palpable infiltrate, discrete papules | Positive |
++ | Erythema, palpable infiltrate, papules, vesicles | Positive |
+++ | Erythema, palpable infiltrate, confluent vesicles | Positive |
The majority of contact allergens has an intrinsic capacity to activate mechanisms of the innate immune response and in this sense can act as irritants. Since this effect is concentration-dependent, using the optimal test concentration of the contact allergen is crucial for the interpretation of the test results. To optimize the performance, test conditions for the most common contact allergens have been standardized. Since relative allergenic and irritative potential of contact allergens can vary a great deal, attempts have been made to classify allergens into those that have a higher irritative than allergenic capacity and those that have a high allergenic and little irritative potency. Calculation of the reaction index (RI) which analyzes the relationship between number of positive patch test reactions and number of questionable or irritant reactions was suggested by the IVDK a parameter to assess the quality of patch test preparations [24, 25]. Similarly, an additional parameter, the positivity ratio (PR) which is defined as the frequency of + reactions among the total number of positive patch test reactions (+ to +++), was developed to be used in combination with the RI to identify problematic allergens [26]. Even though subsequent evaluation of both parameters by a Danish group has challenged the general applicability of this concept [27] it clearly demonstrates that interpretation of the patch test reactions very much depends on the type of allergen tested.
Similarly, false negative reactions may be related to low allergen concentrations in the test preparation or reduced or altered skin permeability. In particular, weak contact allergens that may elicit ACD in sensitive skin areas such as the eyelids, may not be detected when tested on the back skin of the patient. Similarly, weak contact allergens may elicit ACD in areas with a preexisting damage of the barrier function, while they are not strong enough to elicit a reaction on the intact skin of the back. Since the epidermal barrier is crucially involved in determining the level of allergen penetration, modification of the patch test by tape stripping the upper layers of the epidermal stratum corneum prior to allergen application may be used to simulate particularly sensitive or damaged skin areas. Even though this approach has recently been standardized and shown to increase test sensitivity in particular to weak contact allergens [28, 29] interpretation needs to be done very carefully in particular in relation to irritative test reactions which occur more frequently under these conditions.
Both positive or negative test reactions may be re-evaluated by a repeated open application of the suspected substance [30, 31]. Often positive test reactions are observed that are of questionable relevance. They may simply indicate that the patient is sensitized but not necessary allergic to the substance. In this case the substance of interest can be applied twice daily to a 2 × 2 cm area of the cubital skin for consecutive 7 days after which ACD should develop provided that the contact sensitization is clinically relevant. The same approach can be taken when a strongly suspected substance gives a negative patch test reaction or is not available or suitable for occlusive patch testing.
Finally, when photoallergic reactions are suspected a modified patch (photopatch) test may be applied in which two identical panels of allergens are applied to the back skin for 24 h and subsequently one panel is irradiated with UVA (5–10 J/cm2), while the other panel is protected from UV radiation. The test readings are performed at 48, 72 and 96 h according to the same criteria as the standard patch test and allow addressing the role of UV light for the elicitation of the ACD [32].
Treatment of Allergic Contact Dermatitis
Since in the majority of the cases ACD presents as a localized skin reaction and the inflammatory skin infiltrate is accessible to topical treatment, the use of topical glucocorticosteriods applied to the affected skin areas is the mainstay of ACD treatment. In cases of extended skin involvement or generalized ACD short term systemic therapy with glucocorticosteriods may be considered. The topical treatment of ACD corresponds to treatment of other forms of dermatitis. The vehicles used for topical glucocorticosteriod treatment should be adapted to the clinical presentation and state of the eczematous skin reaction. Acute dermatitis requires treatment with hydrophilic creams and/or lotions that provide a cooling and anti-inflammatory effect and help to dry acute weeping or blistering skin lesions. In contrast, chronic dermatitis is usually characterized by dry and brittle skin with lichenification and hyperkeratosis and thus requires more lipophilic vehicles such as lipophilic creams or ointments. In cases of prominent hyperkeratosis in particular on palms and soles the keratolytic effects of salicylic acid or urea may be used to reduce the hyperkeratosis and thus allow better access of the topical anti-inflammatory agents. In case of dermatitis with bacterial superinfection, anti-infective agents such as octinidine or polyhexanide may be used. Supportive measures should include minimizing irritative insults to the skin such as chronic exposure to wet conditions at the work place or repeated hand washing. Since allergic sensitization and elicitation of ACD requires for a variety of allergens some sort of adjuvant irritative effect or skin damage as cofactor to fully develop, reduction of this kind of aggravating cofactors will help to improve the skin condition and to avoid relapses. The mainstay of the treatment of ACD is the anti-inflammatory therapy with topical glucocorticosteriods in the correct vehicle. Topical calcineurin inhibitors such as tacrolimus or pimecrolimus can also be effective, and due to an almost absent induction of skin atrophy may be considered in chronic patients that already have steroid-damaged skin. In patients in whom steroid therapy is not appropriate, UVB or PUVA phototherapy may be an effective alternative. In particular in patients with localized chronic hand dermatitis crème PUVA therapy has proven to be a valuable and well standardized therapeutic option.
However the treatment can only be successful if the offending allergen is avoided. So great care should be taken to identify the relevant allergen and large efforts must be taken to explain the necessity of allergen avoidance. In particular in work related contact allergies this may mean leaving the workplace for good and/or changing the profession. The best prophylaxis of ACD is the complete avoidance of the contact allergen, which in many cases is not possible. In addition, risk factors to develop ACD, such as chronic skin damage that leads to a reduced skin barrier function, should be avoided and/or treated. In the same line of thought, consequent basic skin care using rehydrating lipophilic creams or ointments should be recommended to prevent impairment of barrier function. In addition, the use of barrier creams can be recommended for some allergens and irritants that prevent or reduce penetration of the offending agent.
Finally, animal experiments suggest that in ACD specific tolerance can be induced. Attempts to induce contact allergen specific tolerance in humans have been reported, however only for certain plant allergens like poison ivy [33]. Overall the effects were rather transient and the clinical benefit was not convincing. Similarly oral tolerance induction to nickel has been reported that caused an amelioration of skin manifestation, a reduced skin test reactivity to nickel and a reduced T cell reactivity upon nickel restimulation [34]. Again effects were rather moderate and transient in nature. In summary, so far no effective and lasting allergen specific tolerance induction has been established in humans.
Risk Factors Predisposing to ACD
Many factors may contribute to the susceptibility to ACD. Up to now, genetic studies have not revealed an association of ACD with specific HLA alleles. However, several gene polymorphisms have been identified [35]. These are found in genes regulating skin barrier function, detoxification, innate inflammatory immune responses and T cell responses. Several polymorphisms in the stratum corneum protein filaggrin (FLG) have been found to impact its function and to impair the barrier function of the skin [36]. FLG mutations have been associated with increased susceptibility to atopy [37, 38] but also to ICD and ACD [39, 40]. FLG-deficient mice exhibit increased antigen penetration and exacerbated CHS responses triggered by the irritant croton oil or the contact allergen DNFB [41].
Chemistry and Contact Dermatitis
Contact allergens are low molecular weight chemicals that share one characteristic feature: they are protein-reactive. Due to their small size the chemicals per se cannot be recognized by the immune system and are therefore also designated haptens (half-antigens). Their protein binding is essential for their immunogenicity and antigenicity. Organic chemical allergens can covalently bind to proteins, and metal ions form complexes with proteins. Some contact allergens are not reactive haptens but are pre-haptens which require oxidation or pro-haptens which require metabolic conversion to full haptens. Such chemicals are highly problematic in terms of contact allergen identification in patch testing and in in vitro assays since an adduct and not the parent compound causes sensitization and ACD [42].
A detailed understanding of the relation between the physico-chemical properties, structure and reaction mechanisms and their biological activity is used to develop in silico prediction methods to identify potential contact sensitizers in so-called quantitative structure-activity relationship (QSAR) approaches. Grouping according to so-called mechanistic domains [43, 44] is being analysed with respect to allergenicity and allergenic potency. In a recent study allergenic potency as determined in the mouse local lymph node assay (LLNA) could be correlated with mechanistic domains. The results of that study showed that the more potent contact allergens triggered a broader range of signaling pathways than the less potent ones [45]. These data are encouraging further investigations using QSAR and mechanistic domains to promote our understanding of the relation of the chemistry of contact allergens and its impact on the immune system.
Functional Consequences of Protein Modification by Contact Allergens
The most fascinating question that remains to be solved is how the chemical reactivity of contact allergens is translated into biological responses that can lead to the development of ACD. The interaction of chemicals with biomolecules can alter their function. Since contact allergens are generally chemically reactive electrophiles or complex-forming metal ions, it is to be expected that this reactivity is responsible for their action as allergens. In fact, neutralizing the reactivity of the strong contact allergen 2,4-dinitrochlorobenzene (DNCB) by coupling it to lysine abrogates its ability to induce CHS (our unpublished data). Hypothetically, contact allergens may mimic or interfere with conventional post-translational protein modifications [46].
The chemical reactivity of contact allergens regulates immunity at two levels: the first level is the induction of signaling cascades due to chemical protein modification and the second, resulting level is the regulation of gene expression. Information regarding the identity of the functionally relevant chemically modified proteins is scarce. In one study it was demonstrated that treatment of the human monocytic leukemia cell line THP-1 with the contact allergen DNFB does not modify all cellular proteins but only some which are not necessarily the most abundant proteins [47]. Thus, there is selectivity in the targeting of proteins by contact allergens. New studies begin to analyse contact allergen-modified proteins in 3D skin models. In a recent study high resolution magic angle spinning (HR-MAS) nuclear magnetic resonance (NMR) spectroscopy was used to identify protein modifications by a 13C-labeled electrophile in reconstructed human epidermis (RHE) [48]. Compared to in vitro modification of human serum albumin (HSA) which took several days to be detectable, the in situ protein modification in RHE was detectable after less than 24 h. The predominant lysine modification of HSA, as observed in vitro, was not detected in RHE. This method will be useful to identify contact allergen-modified adducts formed in the skin. Eventually this technique may promote the identification as well as quantitative and qualitative analysis of the contact allergen-modified proteome.
Up to now, only very few proteins, whose modification by contact allergens induce a biological response, have been identified (Fig. 23.3). Human TLR4, the receptor for lipopolysaccharide (LPS, endotoxin) from the cell wall of Gram-negative bacteria, is complexed and dimerized by the metal ions Ni2+, Co2+ [49, 50]. Pd2+ also interacts with human TLR4 [51]. This results in signal transduction for production of inflammatory mediators in the absence of LPS. The cytosolic, cysteine-rich sensor protein for oxidative or electrophilic stress, Keap1, contains cysteine residues that can be modified by contact allergens. This triggers expression of anti-oxidant phase 2 response- and immune genes due to their activation by the Keap1-regulated transcription factor Nrf2. The promoters of these genes harbor an antioxidant response element (ARE). This pathway limits contact allergen-triggered inflammation. Mice lacking Nrf2 have a much lower sensitization threshold for contact allergens, most likely due to an increased level of pro-inflammatory oxidative stress [52, 53]. A third target for modification by electrophilic contact allergens is the ion channel transient receptor potential ankyrin 1 (TRPA1). TRPA1 also harbors cysteines that are attacked by contact allergens such as cinnamic aldehyde or DNCB [54, 55]. Extracellular Ca2+ can then enter the cell. TRPA1 is expressed on a subset of nociceptive nerve fibers, also in the skin, but also on keratinocytes and endothelial cells. The TRPA1 channel is involved in pain but also in itching in inflammatory immune responses [56]. For atopic dermatitis it has been shown that TSLP produced by Th2 cells acts on sensory neurons in the skin to cause itch in a TRPA1 dependent manner [57]. TRPA1-deficient mice show reduced edema, inflammation and itching in cinnamic aldehyde-induced edema in mouse ear skin or in CHS to oxazolone and urushiol [58]. Interestingly, one of the endogenous agonists of TRPA1, 4-hydroxy-2-nonenal (HNE), is produced as a consequence of ROS-mediated oxidation of membrane phospholipids [59]. ROS are important inflammatory mediators in CHS [60].
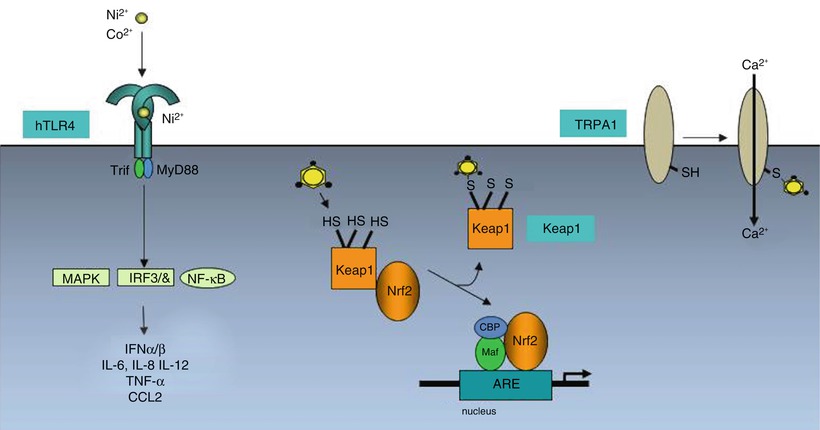
Fig. 23.3
Direct activation of signaling cascades by contact allergens. The metal ions nickel and cobalt form complexes with conserved histidines in human TLR4 resulting in TLR4 dimerization and signaling via NF-kB and MAP kinases and production of inflammatory mediators. Organic chemical allergens such as TNCB or DNCB bind covalently to cysteine residues in the cytosolic protein Keap1. This leads to release of the transcription factor Nrf2, its nuclear translocation and transcriptional activation of the expression of genes containing antioxidant response elements (ARE). These contact allergens can also bind to cysteines in the calcium channel protein TRPA1. Calcium influx activates pathways involved in itch and inflammation
Innate Immune Responses in Allergic Contact Dermatitis
The induction of ACD requires the priming of contact allergen-specific T cells. This depends on two crucial events: activation of the innate immune system and formation of T cell epitopes (Fig. 23.1). It is a peculiarity of contact allergens that they have this dual function [46]. The activation of the innate immune system results in skin inflammation. The most important outcome of this innate inflammatory response in the sensitization phase is the activation and polarization of skin DCs allowing their migration to the draining lymph nodes and presentation of contact allergen to T cells in the context of MHC molecules. Naïve, contact allergen-specific T cells are then primed and polarized towards a Tc1/Th1, Tc17/Th17 phenotype. In the elicitation phase, the innate inflammatory response results in the secretion of cytokines and chemokines, up-regulation of adhesion molecules on endothelial cells and eventually the recruitment of effector or memory T cells into the skin where they exert their effector function to produce ACD.
Recent studies in the mouse CHS model have uncovered mechanistic details of the cellular and molecular innate immune response [22, 61, 62]. The emerging picture clearly indicates that the immune system reacts to contact allergens as if they were infectious agents. This is due to the fact that contact allergens activate the same anti-infectious immune response mechanisms as viruses and bacteria.
The innate cellular response to contact allergens is initiated in the epidermis by activation of keratinocytes and Langerhans cells. Here, the innate inflammatory response is essential to abrogate the naturally tolerogenic milieu in the skin as a major immunologic barrier. As a consequence the normally tolerogenic Langerhans cells are switched to an immunogenic phenotype allowing lymph node migration and T cell priming [63, 64]. Despite the fact that Langerhans cells are dispensable for CHS in some experimental models which usually use saturating contact allergen concentrations, they most likely play a role in ACD under physiological conditions, especially at low contact allergen concentrations and absence of spreading to the dermis [65–67]. Further analysis of the orchestration of the cellular innate immune response has identified mast cells as important initiators of skin inflammation [68]. Mast cell deficiency or mast cell depletion before sensitization significantly reduces CHS in mice. This is due to a role of histamine from mast cells in increasing the permeability of the blood vessels in the skin. The absence of mast cells decreases the infiltration of neutrophils and the emigration of DCs from the skin. Neutrophil depletion efficiently abrogates CHS as does the depletion of subsets of skin DCs [66, 67, 69–71]. Both the sensitization and elicitation phase of CHS depend on the presence of neutrophils [71]. The fact that depletion of one or the other cell type has a similar outcome underlines the essential collaboration of the different innate immune effector cells. The same principle is seen in the orchestration of the molecular mechanisms of contact allergen-induced innate immune responses [46].
Pathogens are recognized by the innate immune system via so-called pattern recognition receptors (PRRs) which are triggered by pathogen- or microbe-associated molecular patterns (PAMPs, MAMPs). Examples for families of PRRs are the Toll-like receptors (TLRs), transmembrane proteins in the plasma membrane or endosomal membranes, the cytosolic NOD-like receptors (NLRs), the RIG-I like receptors (RLRs) and C-type lectin receptors (CLRs) in the plasma membrane [72].
PAMPs/MAMPs can be bacterial cell wall components, flagellin, viral and bacterial nucleic acids, lipids and carbohydrates. These are perceived as danger signals by PRRs and induce innate anti-infectious immune responses. Contact allergens can also trigger PRRs by direct or indirect mechanisms [61]. Nickel- and cobalt ions bind to conserved histidine residues of human TLR4 inducing dimerization and signaling [49, 50]. The murine TLR4 lacks these histidines and therefore is not triggered by Ni or Co. This results in resistance of mice to CHS to these metal ions. However, replacement of the murine TLR4 by the human TLR4 in transgenic mice renders these mice susceptible to CHS [49]. Up to now this is the only known case where contact allergens are directly activating a PRR. For other contact allergens, organic molecules such as 2,4,6-trinitrochlorbenzene (TNCB) or 2,4-dinitrofluorobenzene (DNFB) or oxazolone an indirect activation of PRR has been demonstrated [60, 73, 74]. These contact allergens induce endogenous danger signals. A rapid induction of ROS and release of ATP from skin cells has been revealed [60, 74]. Moreover, the degradation of the extracellular matrix (ECM) component hyaluronic acid (HA) results in fragments that can activate TLR2 and TLR4 [60]. Constitutive overexpression or induction of expression of human hyaluronidase 1 in mouse skin under control of the K14 promoter resulted in lack of CHS when expression was induced before sensitization, most likely due to DC depletion from the skin. CHS was enhanced when overexpression was induced at the time of sensitization [75]. HA breakdown resulted in the emigration of DCs from the skin. The HA effects were dependent on TLR4. These data show a TLR4-dependent role of HA fragments for the mobilization of DC from the skin. The pro-inflammatory role of HA breakdown is evident in Shar-Pei dogs who accumulate HA over time due to an overexpression of hyaluronan synthetase (HAS) 2. Periodic breakdown of accumulated HA results in a periodic fever syndrome in these dogs [76]. These findings illustrate the important role of the ECM in innate immunity [77].
The endogenous danger signal high mobility group box 1 (HMGB1) is involved in the in vitro induction of IL-18 in the human keratinocyte cell line NCTC2544 [78]. HMGB1 was released upon treatment of the cells with the contact allergens para-phenylene diamine (pPD), DNFB or citral.
While ATP triggers the activation of the cytosolic NLPR3 inflammasome that induces the maturation of immature pro-IL-1β and pro-IL-18 via caspase-1, ROS contribute to skin inflammation for example by triggering oxidative HA breakdown and by promoting TLR signaling and inflammasome activation. Nickel also activates the NLRP3 inflammasome [79].
A recent study [80] demonstrated that mice lacking MyD88 or CARD9 are resistant to CHS to TNCB. Irritant CHS to SLS was normal. While MyD88 is an adaptor protein involved in TLR/IL-1R signaling, CARD9 is an adaptor that plays a role in the signaling of CLRs. The study clearly showed that the contact allergens TNCB/TNBS, DNFB and oxazolone can trigger ITAM-Syk-Card9/Malt10 signaling in DCs in vitro resulting in IL-1α and IL-1β production. Signaling was dependent on the ITAM containing adaptor protein DAP12. Syk phosphorylation and CARD9/Bcl-10 mediated NF-kB activation resulted in the production of immature pro-IL-1α and -IL-1β. ROS formation and ROS mediated NLRP3 inflammasome activation for the production of mature IL-α and IL-1β in DCs was Syk-dependent but independent of CARD-9/Bcl-10. The DC-mediated priming and differentiation of contact allergen-specific IFN-γ and IL-17-producing effector T cells was dependent on IL-1β and MyD88. It remains to be determined if and which receptor (e.g., a CLR), couples to this signaling pathway and how it is activated by contact allergens. Moreover, the molecular mechanisms of crosstalk of this signaling pathway with the TLR pathway remain to be determined.
Further important signaling pathways have been identified by genomic or proteomic profiling studies using human cell lines. Two prominent pathways are the aryl hydrocarbon receptor (AhR) and the Keap1/Nrf2 pathway [81]. The AhR is a transcription factor that regulates not only detoxification pathways but also immune processes such as Langerhans cell function and Th17 differentiation. Dietary and endogenous ligands have been identified that modulate immune function [82] and evidence for contact allergen-mediated activation of the AhR has been provided [83].
Disturbed epidermal homeostasis involving keratinocyte death promotes skin inflammation [84] Here, the NF-kB pathway plays an important regulatory role. Mice with an epidermis-specific loss of the NF-kB subunit RelA did not develop spontaneous inflammation. However, DNFB- and oxazolone-induced allergic CHS was aggravated while croton oil-induced irritant CHS developed as in wild type mice [85]. The loss of RelA leads to up-regulation of the small calcium-binding proteins S100A8/A9. These proteins are up-regulated in inflammatory and autoimmune diseases and can have pro-inflammatory, but also anti-inflammatory activity by acting as ligand for TLR4 [86–88]. Increased keratinocyte apoptosis and up-regulation of XIAP associated factor 1 (XIAF1) were observed for contact allergens and croton oil. XIAF1 blocks the anti-apoptotic function of X-linked inhibitors of apoptosis (XIAPs). It was speculated that the selective aggravation of contact allergy may be due to an effect of the contact allergen-specific T cell response on the proliferation of keratinocytes. The nuclear hormone receptor peroxisome proliferator activated receptor (PPAR)-α is involved in the regulation of keratinocyte proliferation and differentiation as well as inflammation. PPAR-α is expressed in keratinocytes but also in Langerhans cells, mast cells and T cells. Its activation can inhibit NF-kB activation [89]. Topical treatment of mice with PPAR-α ligands such as clofibrate counteracts keratinocyte hyperproliferation [90] and reduces TPA-induced irritant CHS and oxazolone-induced allergic CHS. This correlated with a reduction in the levels of TNF-α and IL-1-α [91]. PPAR-α-deficient mice had exacerbated CHS which was associated with impaired IL-2 production in lymph nodes and a decrease in regulatory T cell (Treg) numbers and function [92]. Interestingly, an endogenous PPAR-α ligand, palmitoyl ethanolamide (PEA), is up-regulated along with PPAR-α by contact allergens [93]. These findings underline the importance of dysregulated keratinocyte homeostasis for inflammatory skin diseases.
Many of the mechanisms described above for contact allergens can also be triggered by chemically reactive drugs or drug metabolites. N-acetyl-p-benzo-quinoneimine (NAPQI), the toxic metabolite of acetaminophen is involved in drug-induced liver injury. It can damage hepatocytes. These release self-DNA which acts as DAMP and activates TLR9 on sinusoidal endothelial cells. ROS and ATP then contribute to activation of the NLRP3 inflammasome [94, 95]. Moreover, NAPQI can also activate TRPA1 [96].
The polarization of the cytokine profile secreted by skin DCs is an essential step in the development of ACD. Polarization of T cells towards a Th1/Tc1 phenotype requires IFN-γ and IL-12 or IL-18, IL-21 and IL-27 whereas the polarization of Tc17/Th17 cells requires IL-6 and TGF-β or IL-6, IL1-β and IL-23 [97]. The innate immune response is certainly instrumental in this polarization process. However, there is also evidence for chemical-intrinsic properties that contribute to that [98, 99]. It was observed that the contact allergens DNFB and DNCB induced a type 1 cytokine profile (IFN-γhi, IL-4/-5/-10lo) in the skin draining lymph nodes following topical exposure of Balb/c mice. In contrast, the respiratory allergen trimellitic anhydride (TMA) as well as FITC and DNBSCl induced a type 2 profile (IL-4/-5/-10hi, IFN-γlo). Interestingly, these cytokine profiles correlated with the modification of proteins. In vitro studies using human U937 monocytes showed preferential modification of cellular proteins by DNFB and DNCB, but preferential modification of serum proteins by the other chemicals [100].
A modulation of the epigenetic regulation of gene expression has also been discussed in the context of T cell polarization [101]. A first study analyzed genome wide changes in the methylation of DNA from skin draining lymph nodes of Balb/c mice exposed to DNCB or TMA [102]. Characteristic changes were found for both chemicals with differently methylated regions in various pathways including cytokine and chemokine genes. Future studies using T cells or DCs are needed to identify potential methylation signatures that are specific for contact or respiratory allergens.
Mechanisms of Irritant Contact Dermatitis
Unlike ACD, ICD is caused by chemicals that are not covalently binding to proteins or form complexes with proteins like metal ions do. Chemicals such as detergents, acids, bases and solvents with a variety of physico-chemical properties cause a toxic-irritant skin eczema that may evoke pathologically relevant stress or damage to the skin barrier (Fig. 23.1) [103]. Innate inflammatory immune responses also play a role in ICD but adaptive immunity is not involved. The underlying cellular and molecular mechanisms are not well understood. From the CHS model it is known that ICD to the irritant croton oil is absent in mice lacking TLR4 and IL-12Rβ2 or TLR2 and TLR4 (our unpublished data) but is normal in mice lacking P2X7R [74]. SLS induced CHS is normal in mice lacking MyD88 or CARD9 [80]. Addition of croton oil to sub-sensitizing doses of TNCB or oxazolone is not able to compensate the lack of sufficient innate immune stimulation in the CHS model [104]. On the other hand, addition of SLS to the tolerogen/weak contact allergen restores IL-1β production and prevents 2,4-dinitrothiocyanobenzene (DNTB)-mediated tolerance induction to DNFB [105]. These data suggest that there are contact allergen-specific signaling pathways that cannot be triggered by some irritants. In addition, other pathways are triggered by irritants and there may also be irritant-specific pathways not triggered by contact allergens. Due to the essential irritant effect of contact allergens that is required to induce skin inflammation, it is not surprising that there is an overlap of cytokine and chemokine profiles induced by irritants and contact allergens [106, 107].
Differences between contact allergens and irritants are for example the selective up-regulation of CXCR4 on Langerhans cells by contact allergens. This leads to chemokine-selective migration of LCs to the dermis: LC migration in response to contact allergens is driven by CXCL12 while migration in response to irritants is driven by CCL2 and CCL5 derived from dermal fibroblasts [108, 109]. A recent study revealed a role for basophils in attracting eosinophils in a mouse model of croton oil-induced ICD [110]. Eosinophil-deficient mice had impaired, IL-5-transgenic mice exacerbated ICD. In an in vitro co-culture model basophils secreted IL-4 and TNF-α, and promoted CCL11 expression from fibroblasts. These data suggest a role for basophils in the maturation and attraction of eosinophils to the skin in ICD. Their contribution to production of pro-inflammatory ROS was discussed. Interestingly, basophils and eosinophils are found in other inflammatory human skin diseases including ACD [111–113].
These findings highlight common principles for the immune response to chemicals. Contact allergens, protein-reactive drugs or their reactive metabolites and irritants cause tissue stress and damage. This leads to oxidative stress and the formation of DAMPs, release of DAMPs from stressed and damaged cells and the activation of downstream signaling events that are in part mediated by PRRs. The chemical reactivity of contact allergens and protein-reactive drugs causes the chemical modification of proteins which can result in the direct activation of signaling cascades as is the case for human TLR4, Keap1 and TRPA1 and in in the formation of T cell epitopes. The former leads to innate immune responses resulting in xenoinflammation which is essential for the subsequent activation of the adaptive immune system and the generation of contact allergen- or drug-specific effector and memory T cells [22].
Heterologous Innate Immunity
Contact allergens and irritants are rarely encountered as pure, single substances. Consumer products such as cosmetics, household products or occupational chemicals such as paints or metal cutting fluids often contain combinations of irritants and contact allergens with other chemicals. This combination is of great relevance. The interaction of the different chemicals may result in enhanced skin penetration or augmentation of sensitization and challenge reactions. Facilitated sensitization may be the result [114–116]. Examples are the augmentation effects by combinations of irritants and contact allergens or several contact allergens [117, 118]. Mechanistically, this can be explained based on the specificity of the innate immune response. Due to the activation of identical signaling pathways by different TLRs or other PRRs, a given contact allergen can generate signals for example via TLR4 and these may be amplified by irritants or other contact allergens that also trigger TLR-dependent inflammation. The result is a T cell response and ACD to this contact allergen. If the autologous innate signals triggered by the contact allergen that elicits the T cell response are too weak, augmentation by heterologous innate immune stimulation by irritants or other contact allergens is possible (Fig. 23.2). Moreover, such heterologous innate stimuli may replace missing autologous stimulation. Heterologous innate immune stimulation can also be provided by infections. This may even break tolerance or abrogate genetically based resistance to contact allergy as shown in the CHS model. Here mimicking an infection renders CHS-resistant TLR4/IL-12Rβ2 deficient mice susceptible to CHS. Injection of contact allergen-modified DCs from these mice fail to induce sensitization in wildtype mice. Stimulation of their TLR9 with CpG-oligodeoxynucleotides (CpG-ODN) in vitro restores their sensitizing potential [73]. Likewise, CpG-ODN injection of the CHS-resistant mice at the time of sensitization with contact allergen also abrogates resistance (our unpublished data). Combining nickel which does not trigger mouse TLR4 with LPS allows to efficiently sensitize mice for CHS to nickel [119] and amplifies patch test reactivity [120]. These data highlight the importance of heterologous innate immune stimulation as a process that significantly impacts the outcome of immune responses to chemicals depending on their context [23].
Adaptive Immune Responses in ACD
ACD develops in two phases. The first, sensitization phase is initiated upon skin contact with a chemical allergen. The contact allergen penetrates into the skin and due to its reactivity binds to extracellular, plasma membrane-associated and intracellular proteins. T cells specific for organic chemical contact allergens such as TNCB recognize hapten-modified peptides on MHC molecules [121]. Metal ions such as nickel are recognized by T cells due to complex formation of the metal ion with histidine residues in the MHC molecule and the T cell receptor. For some T cell clones one coordination site is a histidine residue in the peptide bound to the MHC molecule, for others this not the case [122, 123]. The effector T cells in ACD are CD4+ and CD8+ T cells. In the mouse CHS model the effector T cells are usually cytotoxic CD8+ T cells that produce IFN-γ. Moreover, CD8+ IL-17 producing T cells play a role in ACD [113, 124, 125]. In the mouse CHS model a role for dendritic epidermal T cells (DETC) as producers of IL-17 has been described [126] and the ASK1/p38 MAP kinase pathway was shown to be involved in IL-17 production in the elicitation phase of CHS [127]. In human ACD there is some evidence for early infiltrating CD8+ T cells that may cause initial damage. This is similar to results from atopy patch tests [128]. CD4+ T cells are then also detected later. Other effector cells in ACD are infiltrating NK cells that amplify the response due to their IFN-γ production [129]. In the CHS model evidence for contact allergen-specific NK cell responses and solely NK cell-mediated CHS-like reactions in T-cell deficient mice has been provided [130, 131]. However, these reactions seem to be quite different from T cell-mediated CHS [132].
Down-regulation of the immune response in ACD is determined not only by effector T cell death but also by regulatory immune cells such as Treg. In the CHS model ICOS + CD4 + CD25 + Foxp3+ Treg have been identified and a critical role for Langerhans cells in Treg induction has been identified [63, 133]. Invariant NKT cells (iNKT cells) also have regulatory function in CHS [134]. More recently, PU1 + CD4+ Th9 cells have been isolated from ACD skin biopsies of nickel allergic patients [135]. They may have a regulatory role in ACD by acting on Th1 cells directly or via enhancement of IL-4 production by Th2 cells. Interestingly, IL-9 was increased after skin exposure to nickel, rubber and fragrance in a gene expression profiling study of skin biopsies from allergic patients [136].
Tolerance Induction to Contact Allergens
Induction of allergen-specific tolerance is a major goal of the immunotherapy of allergic diseases. Hyposensitization can re-establish allergen tolerance for some years in type I allergies for example to insect venoms, house dust mite and pollen allergens. Immunotherapy (IT) involves the application of increasing doses of the allergen via the subcutaneous (SCIT) or sublingual (SLIT) route. Allergen peptides or recombinant allergens are now also used [137]. The underlying mechanisms involve a shift in the balance between allergen-specific Th2 cells and Treg as well as other regulatory cells such as regulatory B cells, including the de novo induction of such regulatory cells [138]. For ACD there are up to now no established protocols to induce contact allergen-specific tolerance and studies in the CHS model show successful tolerance induction only before sensitization. The clinical problem is therefore not solved, yet [139]. In the CHS model, low zone tolerance (LZT) has been studied for many years. LZT is induced before sensitization and results from the repeated application of contact allergen at doses 100- to 1000-fold below the dose required for sensitization. Recent work has revealed that tolerogenic CD11c + DCs induce contact allergen-specific CD8+ Treg [140]. IL-10 producing CD4 + Foxp3+ Treg were essential for LZT induction and rendered CD11c + DC tolerogenic by direct cell-cell contact via gap junctions. In addition, in the skin draining lymph node, DCs produce TNF-α, which induces the death of effector T cells [141].
The tolerogenicity of contact allergens can be dose-related as in the case of LZT or intrinsic as in the case of DNTB which is a very weak contact allergen and used as a tolerogen. The common principle is most likely the lack of induction of a productive innate immune response. This fails to overcome the homeostatic immunoregulatory default which maintains tolerance and induces active contact allergen-specific tolerance involving DCs and regulatory T cells. The latter occurs due to the fact that T cell epitopes can still be formed by low dose contact allergen or weak contact allergens/tolerogens such as DNTB. The contact allergen is then presented on immature/tolerogenic DCs which results in the induction of CD4+- or CD8+ Treg and the induction of effector T cell anergy and death [63, 140, 141]. The central importance of the innate immune response in shifting the balance between tolerance and immunity was demonstrated by the fact that the irritant SLS was able to prevent tolerance induction by DNTB [105]. The combination of SLS, a heterologous innate immune stimulus [23], with DNTB induced IL-1β. DNTB alone failed to do so.
These findings clearly show that the magnitude of the innate inflammatory immune response is a critical determinant of tolerance and immunity and, most likely, of allergenic potency [142]. It remains to be tested whether tolerogenic adjuvants may be successful as negative heterologous innate stimuli in the re-establishment of tolerance to contact allergens [23]. A major issue is the tolerization of effector and memory T cells. A combination of anti-inflammatory therapies and strategies targeting effector/memory T cells and inducing contact allergen-specific regulatory T cells should be promising.
Biomarker Identification, Gene Signatures
The identification of changes in gene and protein expression induced by contact allergens and irritants will provide important information regarding the mechanisms of action of these chemicals. Pathway analysis can then be used to validate the pathologically relevant pathways and to identify drug targets for new, causative therapies. In addition, characteristic gene signatures can be identified that allow identification of contact allergens and their discrimination from irritants. The classification of chemicals based on physico-chemical and reaction-mechanistic characteristics will reveal whether the gene and protein expression profiles segregate with these characteristics. Dose-response studies will also be important in this context in order to understand the regulation of the balance between immunity and tolerance by contact allergens.
A recent study has provided such initial results from the genomic profiling of patch test biopsies for nickel, fragrance and rubber [136]. One hundred forty-nine genes were commonly regulated by all contact allergens compared to petrolatum as control. Differences between the allergens were observed for their efficiency in the induction of innate immunity and Th1/Th2/Th17/Th22 responses. Another recent genomic profiling study focused on the intra-individual comparison of skin lesion for psoriasis and non-atopic or atopic eczema in patients with both diseases, but also provided data on nickel-induced contact dermatitis [143]. Induced ACD could be differentiated from naturally occuring eczema by the selective down-regulation of late epidermal differentiation markers such as late cornified epithelial (LCE)1 and LCE2 family members, selective up-regulation of adhesion molecules such as ICAM-1 and of extracellular matrix associated HAS3 and epithelial-stromal interaction 1 (EPSTI1) the expression of which is modulated by inflammatory cytokines. Moreover, inflammasome components and neutrophil attracting chemokines were up-regulated in ACD. These studies mark the beginning of future genomic and proteomic studies that will hopefully identify chemical class-specific biomarker signatures for the identification of drug targets, improvement of diagnostics and development of in vitro assays for the identification of contact allergens.
A genome-wide association study (GWAS) with volunteers exposed to the irritants SLS and nonanoic acid revealed differential expression of 883 genes for the two irritants. Only 23 genes were commonly regulated by both chemicals [144].
These data highlight the importance to consider chemical-specific mechanisms. Especially in the case of irritants, it should be rewarding to classify them according to their physico-chemical properties and to analyse their mechanism of action by global technologies as there is little mechanistic understanding regarding the signaling pathways triggered by different irritants. Such studies will not only provide potential therapeutic targets, they will also help to understand the clinically relevant interaction of irritants with contact allergens which may lead to an augmentation of sensitization and of the clinical response, for example due to heterologous innate immune stimulation [23, 117].
In Vitro Assays for Contact Allergen Identification
A great challenge is the replacement of the LLNA (OECD guideline 429) by in vitro assays that identify contact allergens. The LLNA measures the proliferation of mouse lymph node cells following repeated topical application of a test substance or the solvent to the ear skin. Stimulation indices are calculated and effective chemical concentrations needed to give an SI = 3 (EC3 values) are used to classify chemicals including the determination of relative allergenic potency.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


