CHAPTER 6 RARE VASCULAR TUMORS
KEY POINTS
Management of a rare vascular tumor is dependent on the type of lesion.
Rapidly involuting congenital hemangiomas and kaposiform hemangioendotheliomas rarely require surgical intervention.
Noninvoluting congenital hemangiomas, infantile myofibromas, Enzinger intramuscular hemangiomas, and arteriovenous acral tumors are commonly resected.
Surgical intervention for infantile myofibromas, Enzinger intramuscular hemangiomas, and arteriovenous acral tumors is usually required to accurately diagnose these lesions by his-topathologic examination.
Rare vascular tumors are best managed in an interdisciplinary vascular anomalies center.
Several types of rare vascular tumors may be managed by resection. The most frequently encountered are (in order of frequency): congenital hemangiomas, kaposiform hemangioendotheliomas (KHE), infantile myofibromas, arteriovenous acral tumors, and Enzinger intramuscular hemangiomas. Because of the rarity of these lesions, their management will be collectively discussed in this single chapter.
SURGICAL INDICATIONS
The major variable that dictates whether a rare vascular tumor is treated surgically is the type of lesion (see Video 6-1, Rare Tumor). Before surgical intervention is considered, the tumor must be diagnosed. Although more than 90% of vascular anomalies can be diagnosed by history and physical examination, uncommon lesions are more difficult to identify. Consequently, rare vascular tumors are likely to require imaging or biopsy for diagnosis. After the type of lesion is known, its natural history and potential treatments are considered. Some rare vascular tumors regress (for example, congenital hemangiomas and infantile myofibromas), and thus they may not require immediate intervention. KHEs respond to chemotherapy and are seldom treated surgically. Other lesions are managed primarily with excision (for example, Enzinger intramuscular hemangiomas and arteriovenous acral tumors).
SURGICAL MANAGEMENT
CONGENITAL HEMANGIOMA
There are two types of congenital hemangiomas:
Rapidly involuting congenital hemangioma (RICH)
Noninvoluting congenital hemangioma (NICH)
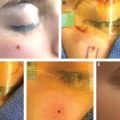
Both types are fully formed at birth and are located on the head/neck (about 45%), extremity (about 45%), or trunk (about 10%). Lesions are red purple, well defined, and usually surrounded by a pale halo. Unlike infantile hemangiomas, congenital hemangiomas do not have postnatal growth. A RICH begins to involute immediately after birth and has completely regressed by approximately 12 months of age. A NICH does not improve and maintains the same appearance.
Rapidly Involuting Congenital Hemangioma
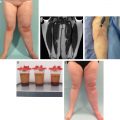
In contrast to an infantile hemangioma, which often leaves behind excess fibroadipose tissue, a RICH can cause subcutaneous atrophy after involution. A patient may have a cutaneous depression or residual telangiectasias that causes psychosocial morbidity. If the skin is relatively normal, adipose atrophy can be reconstructed with fat grafting. Telangiectasias, scarring, or anetoderma of the integument may be improved by excising the area. If the region is large, serial resection might be required. A deformity in a visible location (for example, the face or hands) can be improved with surgical intervention between 3 and 4 years of age, before the child’s self-esteem and long-term memory begin to form. Because most RICHs are located on the trunk and extremities, a residual deformity can usually be camouflaged with clothing. Patients often present in early adolescence for surgical improvement when their appearance begins to be more important to them.
Noninvoluting Congenital Hemangioma
A NICH changes minimally after birth and often causes psychosocial distress. Although laser treatment may be able to improve its color, the skin and deep dermis are abnormal, and thus a deformity will remain. The most common intervention for NICHs is resection, which can require serial excisions if it is large. Similar to RICHs, many are located outside of the head/neck, and patients typically seek intervention in early adolescence to improve their appearance. Congenital hemangiomas are benign, and negative margins in the resection specimen are unnecessary. Lesions are not at increased risk of intraoperative bleeding.
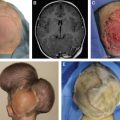
KAPOSIFORM HEMANGIOENDOTHELIOMA
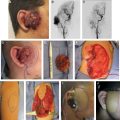
A KHE is a locally aggressive neoplasm that does not metastasize. It affects 1 in 100,000 persons, and 50% are present at birth; the rest usually develop in early childhood. KHEs appear reddish purple and do not have well-defined borders. Lesions affect the head/neck (40%), trunk (30%), or extremity (30%). Seventy percent cause life-threatening thrombocytopenia (Kasabach-Merritt phenomenon). KHEs typically undergo partial regression in early childhood but do not completely involute. Patients can develop contractures and chronic pain from fibrosis. Surgical treatment is rare for two reasons: (1) Lesions are almost always diffuse, involving multiple tissue planes, and (2) effective pharmacotherapy is available.
During infancy patients usually are treated with either intravenous vincristine or oral sirolimus to shrink the tumor, treat thrombocytopenia, reduce the risk of chronic pain, and prevent contractures from fibrosis. Surgical intervention for KHEs is reserved for: (1) localized lesions that are amenable to resection and (2) secondary reconstruction for fibrosis/contractures. Consideration should be given to excision in the rare circumstance that the KHE is localized and amenable to complete (or near-total) resection. Removing the lesion in infancy will obviate the morbidity of systemic pharmacotherapy. If possible, the surgeon should wait until the child is older than 6 months of age to reduce the anesthetic risk. Platelet function must be maximized before surgical intervention; patients may require vincristine or sirolimus before surgery to normalize their platelets.
KHEs do not metastasize and have minimal risk of recurrence after extirpation. Consequently, achieving normal margins is not mandatory. It can be difficult to distinguish the lesion from normal surrounding skin because of edema, fibrosis, and erythema. As much of the tumor as possible should be removed; it is not critical to include normal margins if it would complicate reconstruction. Even a subtotal resection will significantly reduce the patient’s symptoms and is likely to obviate the need for drug treatment.
Surgical intervention for KHEs during the acute phase in early childhood is less frequent than reconstruction of the area for chronic fibrosis. KHEs may leave behind damaged integument that can be improved surgically (for example, contracture, discoloration, and abnormal skin contour). Reconstruction is based on the type of deformity. Contractures are released by local tissue rearrangements and/or skin grafting. Abnormal areas of integument can be serially excised. Tufted angioma is closely related to a KHE and treated with the same pharmacotherapy; the lesion is often localized and thus more amenable to resection.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


