Type of death
Characteristics
Molecular markers
Critical signaling molecules
Apoptosis
Can be induced by extracellular (TNF-α, FasL, TLRs…) or intracellular (ROS, cytochrome-c) stimuli
Initiated by formation of the apoptosome
Considered ‘anti-inflammatory’
Ladder; Blebbing; TUNEL positive; extracellular Annexin-V; Cleavage of: caspase-3, Rip1, iCAD, PARP; Cytoplasmic cytochrome-c; Membrane integrity maintained
TNFR, Fas, TRAIL, TLR4, TLR2, Bax, Bak
↓
FADD, TRADD, MyD88, APAF, other DD mlcls
↓
Caspase-8, −9, −6, −7, −3
Necrosis/Necroptosis
Classical inflammatory cell death
Originally thought to be ‘unordered’, now specific signaling pathways have been identified
Release of PAMPS/DAMPS; Increased TNF-α; Cell swelling; Upregulation of Rip3; Phosphorylation of MLKL
Any apoptotic initiator in presence of caspase inhibitor, DNA damage
↓
Rip1, Rip3
↓
MLKL
Autophagy
Homeostatic process
Can lead to cell death by destruction of cellular components
Involves formation of autophagosome which fuses with lysosome
Is regulated by status of other cell death pathways
Covalent bonding of Atg5 with Atg12; Formation of LC3-II
Beclin, PI3K
↓
LC3-1, Atg family proteins
↓
Lysosomal fusion
Pyroptosis
Initiated by activation of the caspase-1 inflammasome
Dependent on intracellular PRR, many of which require a ‘first hit’ to be upregulated
Leads to inflammatory cell death
Release of PAMPs/DAMPs; Increased IL-1β and IL-18; Cell swelling; Cleavage of caspase-1; TUNEL positive
NLRP1, NLRC4, Aim2, other intracellular PRR
↓
ASC, CARD domain containing adaptors
↓
Caspase-1
Apoptosis
Keratinocyte apoptosis is a classic example of how programmed cell death is utilized to maintain a healthy epidermal skin barrier. As a keratinocyte matures it moves from the basal side of the epidermis towards the apical interface with the environment. During this migration it halts proliferation, undergoes keratinization, and ultimately begins the process of apoptotic cell death so that it can be sloughed off once it reaches the apical surface. This apoptotic cell death can be acutely activated by environmental stress, such as UV-radiation, or can be slowly achieved for homeostatic maintenance of the epidermal tissue. The apoptotic death of these important barrier cells is inevitable and crucial to their function.
There are two classical pathways that, upon activation, lead to an immunologically silent apoptotic cell death, the extrinsic and the intrinsic pathways [16]. The extrinsic pathway is initiated by engagement of cell surface death receptors, most notably TNFR and Fas, that associate with the scaffolding proteins, TRADD and FADD, respectively, to create a death-inducing signaling complex (DISC) [17]. The intrinsic pathway is initiated by release of cytochrome-c from the mitochondria, followed by formation of the apoptosome [18]. Many varied conditions can cause the release of cytochrome-c from the mitochondria, including the build-up of reactive oxygen species (ROS), genomic instability, metabolic instability, and the recognition of any other condition in which it makes more teleologic sense for the cell to die rather than live. The apoptosome is initiated by the binding of cytochrome-c to APAF-1 which is then able to oligimerize to create a circular structure that activates the apoptosis execution proteins named caspases (Fig. 13.1).
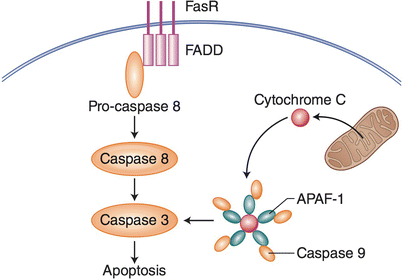
Fig. 13.1
Extrinsic and intrinsic apoptosis. For extrinsic apoptosis, engagement of Fas Receptor by Fas triggers a signal cascade that involves FADD, pro-caspase 8 activation, and programmed cell death by apoptosis. Intrinsic apoptosis can be triggered by intracellular ROS, resulting in the release of mitochondrial cytochrome c, which binds to APAF-1, resulting in executioner caspases that trigger apoptosis
Both pathways, either through a DISC or the apoptosome, result in cleavage of caspase-3 that, in turn, cleaves and activates caspase-activated DNase (CAD). Once activated, CAD translocates into the nucleus where it starts digesting genomic DNA, among other cellular components, which is released from the cell in apoptotic vesicles. This digestion process results in the “DNA laddering” that is a hallmark of apoptotic cells [19]. DNA laddering is the result of DNA cleavage by CAD between nucleosomes, which results in DNA fragments increasing in size consistently by about 180 base pairs, which is the amount of DNA wrapped around a histone. In addition to active cleavage of DNA, apoptotic cells undergo a loss of survival signals and homeostatic processes. Experiments using caspase inhibitors show that without the ability to activate the apoptotic pathway, cell death can be skewed to an inflammatory, caspase-independent cell death that is dependent on the formation of specific signaling complexes and is termed necroptosis [20].
Additionally, ER stress and activation of the associated unfolded protein response is another cause for the initiation of apoptosis. When secretory proteins are not able to fold correctly and emerge from the ER, the unfolded protein response is activated and can lead to activation of caspase-3 and subsequent apoptosis [21]. This is just one example of an instance where a cell cannot survive in its current state and so apoptotic death is initiated so that further damage to surrounding cells is not propagated, or survival of a terminally damaged cell is not prolonged [22]. Apoptosis is an elegant way that organismal homeostasis is able to be maintained at the cellular level.
Necrosis
The term necrosis is pathologically described as irreversible cell damage that leads to the extracellular release of intracellular material and destruction of surrounding tissue [23]. This definition originated mostly from gross observation of what was deemed to be ‘necrotic’ tissue. Such tissue can be separated into five main distinct categories that are based on morphological patterns: coagulative, liquefactive, caseous, fat, and fibrinoid necrosis [24–28]. As molecular biology techniques advanced, the definition of necrosis has evolved to include more biochemically defining characteristics. These include swelling of the cell followed by the release of specific damage-associated molecular patterns (DAMPs), and other pro-inflammatory molecules from dying cells [29]. Necrotic cell death was originally viewed as a ‘back-up’ or default way for a cell to die that simply involved the non-specific swelling and lysis of a cell. More recently several specific signaling pathways that result in necrosis have been identified, making necrosis an umbrella category for ordered cell death that results in a pro-inflammatory lytic cell death. Of the recently described necrotic signaling pathways, a process termed necroptosis has been the most thoroughly described.
Necroptosis is initiated by the Rip1-Rip3 protein complex and leads to inflammatory cell death [30]. The many consequences of and cellular mechanisms leading to the Rip1-Rip3 protein complex, and subsequently necroptosis, is still a widespread active area of investigation. However, to date, the necroptotic pathway has been most completely described downstream of extracellular death receptors, such as TNFR1 (Fig. 13.2). In this scenario, TNF-α engages TNFR1 inhibition of caspase-8 activation by zVAD has been shown to activate the necrosome which leads to a necroptotic cell death. A limited number of markers of necroptosis have been identified, among them an increase in Rip3, and maintenance of intact Rip1, and phosphorylation of Mixed lineage kinase (MLKL) [31]. This is in contrast to Rip1 cleavage which is associated with caspase-8 activation and apoptosis [32, 33]. The recently reported ability of Rip1 to mediate production of TNF-α during necroptosis helps to describe the inflammatory nature of necroptotic cell death and may prove to also be a hallmark of necroptosis [34]. Many inflammatory and necrotic phenotypes are reported as a result of inhibiting the apoptosis pathway components. For example, mice in which caspase 8 is conditionally knocked-out in hepatic cells die of necrosis of the liver [35]. Also, FADD conditional knockout in intestinal epithelial cells leads to a spontaneous colitis phenotype that is rescued when Rip3 is also knocked out [36]. In a MyD88 deficient model it has also been found that necroptosis, rather than apoptosis, is activated in UV-radiated KC [37], suggesting an unexpected role for innate signaling pathways in dictating cell death outcomes. Previously described inflammatory phenotypes may now be better explained as a consequence of using necroptosis as a default death pathway when apoptosis is inhibited.
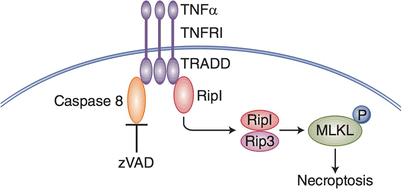
Fig. 13.2
Necroptosis. This form of cell death was originally identified using an extrinsic death pathway TNF-a receptor and its ligand TNF-a. When the activities of executioner caspases are blocked by a chemical such as z-VAD, an alternate, default death pathway becomes active (necroptosis). In this form of inflammatory cell death MLKL is phosphorylated, and migrates to the plasma membrane, resulting in membrane leakiness, and cell swelling and inflammatory death. Necroptotic cell death can occur in naturally occurring circumstances, such as reperfusion injury
Pyroptosis
Pyroptosis is a pro-inflammatory cell death that is the result of the activation of an inflammasome. During pyroptotic cell death many pro-inflammatory zymogens are cleaved and activated by the pyroptosis executer protein, caspase-1 [38]. These zymogens include the extensively studied pro-inflammatory cytokines, IL-1β and IL-18, which lead to the wide spread inflammation associated with this type of programmed cell death [39]. Pyroptotic cell death is classically thought to require a priming step which initiates the production of the aforementioned zymogens and necessary machinery which can subsequently be activated to form an inflammasome [40]. However, more recently it has been reported that inflammasome activation can occur independently of an initial priming step, this is an active area of current investigation (Fig. 13.3) [41].
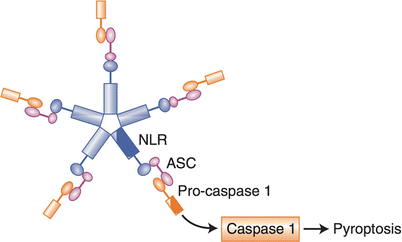
Fig. 13.3
Pyroptosis. This form of cell death is pro-inflammatory, because the inflammasome is activated by NLR, ASC and activation of caspase 1, resulting in the cleavage (activation) of IL-1beta and IL-18, two pro-inflammatory molecules. This form of cell death is activated by intracellular pattern recognition receptors (Nod receptors), which are typically mobilized by PAMPS forming a “wheel of death”, but can also be mobilized by DAMPS (such as extracellular ATP)
The inflammasome, is a cytosolic signaling complex that is dependent on the activation of intracellular pattern recognition receptors (PRR) and is anecdotally referred to as the ‘wheel of death’. To initiate an inflammasome cytoplasmic PRRs identify an agonist and then begin to cluster into a wheel shaped aggregate [42, 43]. This signaling complex continues to recruit adaptor proteins, including apoptosis-associated speck-like protein containing a CARD domain (ASC), that scaffold to eventually include a CARD-domain containing protein [39]. The cluster of Caspase activation and recruitment domain (CARD)-domain containing proteins are then able to activate caspase-1, which directly activates the cell death and severe inflammation associated with pyroptosis. Inflammasome activation is also able to mobilize the adaptive arm of an immune response against the microbes it initially sensed [44]. Mobilization of both the innate and adaptive arms of the immune system combined with the death of the infected cell allows pyroptosis to be an effective way for our body to rid itself of pathogenic microbes.
Autophagy
Autophagy is a homeostatic cell process that can be, but is not always, associated with cell death [45]. This process is initiated when a cytoplasmic fraction is recognized as needing to be destroyed or recycled. The damaged/infected fraction of the cytosol is engulfed into a double membrane organelle termed the autophagasome which then fuses with a lysosome. The lysosomal enzymes then have access to the contents of the autophagosome which are promptly digested. If too much of the cytosolic fraction has been labeled for destruction by autophagasome, the cell can die by autophagy. This autophagic cell death is considered non-inflammatory and almost as ‘silent’ as apoptotic cell death (Fig. 13.4) [46].
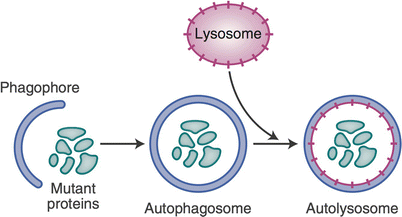
Fig. 13.4
Autophagy. This is a homeostatic process that may or may not result in cell death. Damaged intracellular organelles are engulfed by a membrane termed the autophagosome, which then fuses with a lysosome, resulting in the digestion of the damaged organelle. Limited autophagy does not result in cell death. Extensive organelle damage can lead to cell death: an autophagic, silent, non-inflammatory cell death
Many different conditions can lead to the activation of autophagy all of which have certainly not yet been appreciated. The anti-inflammatory nature of autophagic cell death has been thought to serve several different purposes [47]. It is an inconsequential way of surveying the cytosol of a cell without initiating a robust immune response [48]. It has also been labeled as a way for cells to regulate and dampen inflammatory cell death that would otherwise run rampant during a microbial infection. Activation of autophagy following disease-related oxidative stress can regulate the inflammatory response and widespread inflammation that would normally result from inflammasome activation by ROS [49].
Immune Signaling Resulting in Cell Death
Nod-Like Receptors (NLRs)
NLR signaling is most classically associated with activation of the inflammasome and subsequent pyroptotic cell death. However, in the years following their initial discovery it has been shown that signaling downstream of NLRs can lead to many more types of cell death than simply pyroptosis. For example, interferon-inducible protein AIM2 and NLRP3 inflammasomes have been reported to activate both pyroptotic and apoptotic cell death [50]. In fact, recent research has gone so far as to call NLRs the ‘master regulators of inflammation’ [51]. This title, while perhaps overstated, has some truth to it. NLRs have, to date, been associated with almost all reported types of programmed cell death. NLRs are also able to stimulate an autophagic response upon activation [52–55]. Furthermore, phagocytosis of autophagic cells has been found to activate the inflammasome and cause the production of many pro-inflammatory cytokines including TNF-α, IL-6, IL-8, and IL-1β [56].
However, activation of NLRs seems to be exclusive from necrosis; if a cell has activated inflammasomes, it will not die by necrosis. Lysosome rupture seems to initiate necrosis and control NLR signaling [57]. Importantly, the two main pathways leading to inflammatory cell death do not appear to overlap, which may allow for the organism to be able to recover from inflammation rather than compound it.
Non-classical Signaling Pathways and Mechanisms Controlling Cell Death
As members of the IL-1 family receptors, toll-like receptors (TLR) are classically known to induce inflammation and, similarly to NLRs, lead to the clearance of the pathogen that they sense. However, as more research is done on these extremely dynamic signaling pathways, it is becoming clear that they are capable of producing many more outcomes than initially thought. For example, extracellular TLRs primed during pyroptotic cell death causes different isoforms of damage associated molecular patterns (DAMPs) to be released, which skews the resulting immune response [58]. A pathway in which engagement of TLR2 and TLR4 signaling stabilizes the autophagy promoting protein Beclin-1 has also recently been described [59]. In this example of a non-classical cell death control mechanism, activation of a pro-inflammatory signaling pathway leads to immunologically silent autophagic cell death. This new association could represent an internal check for inflammation, which does not allow TLR-induced inflammatory responses to go out of control.
As previously mentioned, a novel MyD88-dependent TLR4 signaling pathway that directly activates apoptosis in response to UV has also recently been reported. In the absence of a functional TLR4/MyD88 signaling pathway, cell death after UV-irradiation is skewed to a more inflammatory ordered necrotic cell death termed necroptosis. Because TLR4/MyD88-deficient cells are dying in a more inflammatory and immunogenic way, MyD88−/− and TLR4−/− animals are resistant to systemic immune suppression caused by UV-irradiation, which plays a critical role in the development of UV-induced skin cancers. Activated apoptotic mechanisms previously thought to be a less damaging, less inflammatory, and therefore more desirable outcome during infection, may prove to facilitate the propagation of long-term consequences, such as carcinogenesis.
It is now clear that there is much more cross-talk between apoptosis, necrosis, and autophagic signaling pathways than previously appreciated. Signaling proteins once thought to be mutally exclusive are now being shown to play a role in multiple programmed cell death pathways [60]. If we can identify non-classical cell death pathways activated during tissue damaging circumstances, and determine the consequences of them, we may be able to skew toward alternative cell deaths that will allow for a more immunogenic environment, propagation of fewer potentially cancerous mutations, and an overall more favorable outcome.
Cell Death in Skin Diseases
Dysfunctional or altered cell death can result in a variety of different dermatological diseases with varying clinical presentations and characteristics. In many of these diseases, inflammatory responses of the immune system are altered in conjunction with impaired cell death leading to chronic inflammation or immune suppression. Not only is abnormal cell death involved in the pathogenesis of dermatological diseases, but cell death can also be induced using therapeutic techniques to treat a wide range of dermatological diseases (Table 13.2).
Table 13.2
Diseases and therapies associated with different types of cell death
Disease/therapy | Pathophysiology | Related cell death | References |
|---|---|---|---|
Psoriasis | Hyperproliferation of the epidermis T-lymphocyte mediated disorder Associated with comorbid chronic systemic inflammatory diseases (Crohn’s Disease) | Decreased apoptosis | |
Systemic Lupus Erythematosus (SLE) | Antibodies against nuclear autoantigens Malar or discoid rashes, oral ulcers and photosensitivity | Increased apoptosis and decreased clearance of apoptotic bodies; secondary necrosis; increased apoptosis to UV light (photosensitivity) | |
Keloids | Overgrowth of granulation/scar tissue Increased fibroblast activity | Decreased apoptosis | |
SJS-TEN spectrum | Drug hypersensitivity reaction Blistering and widespread epidermal detachment | Increased KC apoptosis; evidence of necroptosis | |
Skin cancer | Tumor formation after chronic UVR exposure | Decreased apoptosis | |
Crohn’s Disease | Chronic inflammation of terminal ileum Abnormal immune response to commensal bacteria of the gut | Necroptosis, Necrosis | |
Ischemic Reperfusion (IR) injury | Tissue ischemia followed by reperfusion that initiates an inflammatory response that may exacerbate local injury and lead to impaired remote organ function MPT-induced necrosis involving cyclophilin D and RIP kinase mediated necroptosis | Necrosis, Necroptosis | |
Graft vs. host disease (GVHD) | Most common complication of BMT Donor T cells recognize host tissue (APC) as foreign | Increased apoptosis | |
Photodynamic therapy (PDT) | Cell death induced by light after sensitizing the tissue with a photosensitizing agent ROS formation after photosensitizing agent exposed to light induces cell death | Apoptosis; necrosis/necroptosis | |
Ingenol Mebutate (Picato gel) | Rapid necrosis of cells in the lesion/tumor followed by neutrophil-mediated, antibody-dependent cellular cytotoxicity of the residual tumor cells | Necrosis | |
Cryosurgery | Direct cell injury followed by vascular stasis and tissue ischemia | Necrosis |
Conditions Associated with Decreased Apoptosis
There are several common skin disorders during which decreased apoptotic cell death causes a wide variety of pathogenic symptoms. These conditions include psoriasis, keloids, and skin cancer.
Psoriasis
Psoriasis is a chronic immune-mediated inflammatory skin disease usually manifesting as erythematous plaques with silvery scales, most often on the elbows, knees, hands, feet and lower back. While skin lesions are the most common manifestation, individuals can also develop psoriatic arthritis and nail dystrophy [61]. During psoriasis pathogenesis, epidermal KC associated with lesions become hyper-proliferative and are resistant to apoptotic cell death. This resistance is due to both the overexpression of intracellular anti-apoptotic molecules and extracellular cytokines which influence cell fate [62, 63].
While psoriasis is classically viewed as a T-lymphocyte mediated inflammatory skin disease, macrophages have also been implicated as having a role in its pathogenesis [64]. Research using a mouse model of psoriasis has shown that elimination of macrophages attenuated psoriatic symptoms, suggesting that psoriasis development depended on macrophages and not necessarily T-lymphocytes. Macrophages are the major source of TNF-α in the skin [65]. Previous studies have demonstrated that TNF and TNFR1 signaling is critical in the development of a psoriasis-like inflammatory skin disease in mice models [66]. Treatment with IL-11 or IL-4, which inhibit macrophage production of TNF-α [67, 68], has shown to improve symptoms in patients with psoriasis [69, 70]. Cells in psoriatic lesions may exhibit abnormal signaling and contribute to the inappropriate activation of the immune system. This deregulation of immune signaling alters the homeostatic apoptosis which is critical to maintaining a healthy epithelial layer.
Keloids
Keloids develop due to abnormal wound healing leading to overgrowth of granulation tissue that enlarges and often extends beyond the margins of the original wound. Keloids do not spontaneously regress and often recur after excision [80]. Keloid formation is believed to be attributed to increased proliferation and decreased apoptosis of fibroblasts [81–84]. Fibroblasts are important for producing and depositing ECM proteins during wound healing. Fibroblasts of normal skin tend to be quiescent with very low activity; however, keloid-derived fibroblasts have higher densities and proliferative activity [85]. Many studies have shown that keloid fibroblasts exhibit reduced apoptotic rates compared to normal fibroblasts [81–84]. These reduced rates in apoptosis have been attributed to alterations in gene expression in both the fibroblasts themselves and the surrounding KC [83, 84, 86–95].
Various immune cells such as T lymphocytes and macrophages have also been implicated in keloid formation since they are involved in coordinating fibroblast activity during wound healing [96–100]. Failure to clear the inflammatory cells at the wound site due to decreased apoptosis may lead to the imbalance of inflammatory cell populations observed in keloid tissue and the increased activity of fibroblasts contributing to keloid formation [101, 102]. Therefore, the decrease in apoptosis observed in keloid tissue seems to account for the persistence of cellular infiltrate and the accumulation and enlargement of granulation tissue at and beyond the wound site.
Skin Cancer
Skin cancer results when there is hyperproliferation and/or decreased apoptosis of mutated cells that form a tumor which progresses to a malignant state, usually after chronic ultraviolet radiation (UVR) exposure. Actinic keratoses (AK), squamous cell carcinoma (SCC), and basal cell carcinoma (BCC) have higher proliferation rates and decreased apoptotic rates compared to normal skin [63]. These differences were most likely associated with increased expression of anti-apoptotic molecules and decreased expression of death receptors that induce apoptosis [225–230]. Mutations in P53, a tumor suppressor gene, are also commonly associated with skin cancers [123–127] and is often induced by chronic UVR exposure, preventing cell cycle arrest or DNA damage-induced apoptosis [128–130]. Consequently, mutated cells are able to survive and clonally expand outside their original compartment eventually forming a tumor [131–133]. Clonal expansion not only requires a decrease in apoptosis of the mutant cells, but also apoptosis of the surrounding cells. Apoptosis of KC adjacent to the mutant cells allows the mutant clones to expand and develop into a tumor [133]. Therefore, it appears that both apoptosis and decreased apoptosis are critical at specific stages in skin carcinogenesis.
In addition to impaired apoptosis, UV-induced immune suppression plays an important role in skin carcinogenesis. Individuals on immunosuppressants have greater incidences of skin cancer on sun-exposed areas compared to immunocompetent individuals [134–136], further supporting the notion that UV-induced immune suppression is critical for the development of skin cancer. UV-induced DNA damage seems to trigger immunosuppression [137–141] by promoting the production of cytokines and apoptosis-inducing ligands like TNF and FasL [130, 142, 143], without which UV-induced immunosuppression does not occur and skin cancer incidence is decreased [144, 145]. Based on this evidence, skin cancer formation involves a state of immunosuppression and decreased inflammatory responses in combination with dysregulated apoptosis to allow the mutated cells to persist and progress into a tumor.
Conditions Associated with Increased Apoptosis
Just as there are several common skin disorders associated with decreased apoptotic cell death, there are many skin conditions with pathogenic symptoms due to increased apoptotic cell death. These conditions include SLE, SJS-TEN spectrum, and GVHD.
Cutaneous and Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease that affects multiple organs including the skin. Individuals with SLE can develop malar or discoid rashes, oral ulcers and photosensitivity [71]. SLE is characterized by the presence of autoantibodies against nuclear antigens. The pathogenesis of SLE is believed to be due to an increase in apoptosis and a decrease in the clearance of apoptotic cells [72–75]. Many studies have shown an increase in KC apoptosis in LE lesions compared to normal controls [76, 77], which may be mediated by increased Fas expression and decreased Bcl2 expression [77]. While the mechanism remains unclear, it appears that clearance of apoptotic cells by phagocytes is impaired in lupus patients and mouse models. Accumulating apoptotic cells that would normally be cleared by phagocytes undergo secondary necrosis, releasing their intracellular contents and exposing their apoptosis-induced autoantigens to the immune system, which present as danger signals to APCs [78]. The antigens are taken up by DC leading to IFN-α production and activation of T and B cells [79]. Antibodies are produced against the autoantigens resulting in chronic inflammation and the development of SLE [79]. Therefore, while there is increased apoptosis which may be associated with immunosuppressive effects, the decreased clearance of the apoptotic cells results in secondary necrosis and the release of intracellular material, and exposure of the immune system to otherwise cryptic self-antigens. This activates the immune system leading to the chronic inflammatory state of SLE.
Severe Drug Hypersensitivity Syndromes
Stevens-Johnson Syndrome (SJS) and toxic epidermal necrolylsis (TEN) are life-threatening skin diseases caused by drugs or infections characterized by blistering lesions and widespread epidermal detachment [103]. SJS involves less than 10 % of the body surface area (BSA) and TEN involves greater than 30 % of the BSA [104]. The pathogenesis of SJS-TEN involves widespread KC apoptosis mediated by cytotoxic T lymphocytes (CTLs) [105]. CTLs may induce KC apoptosis via the perforin/granzyme pathway and/or the Fas/FasL pathway [106–108]. Increased levels of these apoptotic molecules have been found in epidermal KC, blister fluid and serum of TEN patients [106, 109–111]. While FasL on KC have been implicated in inducing KC apoptosis seen in SJS-TEN, soluble FasL (sFasL) from PBMC, not KC, has been suggested as the main mediator of KC apoptosis in SJS-TEN [112]. Whether FasL on KC or sFasL from PBMCs is responsible, evidence suggests that the Fas/FasL pathway is important in KC apoptosis and the pathogenesis of SJS-TEN. This is corroborated by the favorable outcomes observed in vitro and in vivo with administration of anti-Fas antibodies or IVIG, which contains naturally occurring anti-Fas antibodies [111, 113–120]. Along with Fas/FasL and perforin/granzyme pathways, granulysin has also been implicated in the pathogenesis of SJS-TEN [121]. Granulysin, a molecule demonstrated to be cytotoxic in vitro, showed the greatest staining in TEN skin biopsies compared to any other SJS-TEN-associated molecule and granulysin protein levels correlated with clinical severity [121]. This suggests that granulysin may contribute to epidermal destruction in SJS-TEN and may better explain the widespread KC apoptosis since granulysin can induce apoptosis without direct cell contact, unlike the Fas/FasL and perforin/granzyme pathways [121]. While most of the evidence points to apoptosis as the main mechanism of KC death and the development of SJS-TEN, recent studies have suggested that necroptosis may also be involved in the pathogenesis of SJS-TEN. It has recently been reported that severe adverse blistering reactions to topical therapies may be the result of secreted Annexin-V which initiates a necroptotic, rather than apoptotic, cell death [122]. Although the pathogenic mechanisms are still being elucidated, it appears that these drug sensitivity reactions involve an inflammatory response leading to inappropriate KC death and widespread epidermal detachment.
Graft Versus Host Disease
Graft vs. host disease (GVHD) is the main complication following a bone marrow transplant (BMT) [170]. GVHD is an immunological disorder affecting multiple organ systems mainly the skin, liver and gastrointestinal tract. The skin is the most frequently affected and usually the first organ involved. A pruritic, maculopapular rash develops that can spread all over the body typically sparing the scalp. GVHD is thought to progress in three stages. First, the host tissue is damaged as a result of the BMT conditioning regimen before the transplantation, usually with chemotherapy. The damaged host tissue produces proinflammatory cytokines which activate host APCs and upregulate MHC antigens on the APCs [171–182]. Second, donor T cells recognize the antigens on host APC leading to T cell activation, differentiation and migration. This recognition and activation can occur in MHC-mismatched and MHC-matched transplantations [183, 184]. Activated T cells then produce Th1 cytokines (IFN gamma, IL-2, TNF-α) [185, 186], which stimulate CTL. Third, CTL and inflammatory cytokines promote inflammation and tissue injury by inducing apoptosis. CTLs induce apoptosis mainly via the Fas/FasL or perforin/granzyme pathways [107, 108]. In the skin, Fas-mediated apoptosis by CTLs and TNF-α-mediated apoptosis seem to contribute the most to KC apoptosis and tissue damage [187]. Anti-Fas or anti-TNF-α antibodies were shown to diminish GVHD skin lesions [188] and using both anti-Fas and anti-TNF-α antibodies together completely blocked the skin lesions, suggesting that Fas and TNF-α signaling contribute to tissue injury in GVHD [188]. Based on current evidence, GVHD pathogenesis involves an inappropriate inflammatory response resulting in excessive apoptosis and tissue destruction that can affect a variety of organs, including the skin.
Conditions Associated with Necrosis/Necroptosis
Not only are there skin disorders associated with impaired apoptosis, there are many common dermatological conditions linked to necrosis or necroptosis. These conditions include ischemia reperfusion (IR) injury and Crohn’s Disease.
IR injury involves tissue ischemia followed by reperfusion that initiates an inflammatory response that may exacerbate local injury and lead to impaired remote organ function [152]. While it is not well studied in the skin, IR injury has been well studied in the central nervous system and cardiovascular system and the pathogenesis of such injury involves fundamental processes that occur in the skin. IR injury can result after acute vascular occlusions followed by their respective reperfusion strategies, surgical procedures and shock or trauma [153]. Systemic effects of IR injury can include serious conditions like systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS). Majority of the cell death in IR injury is believed to be attributed to necrosis [154, 155]. Various types of necrosis have been linked to the cell death seen in IR injury such as MPT-induced necrosis involving cyclophilin D [156], RIP kinase-mediated necroptosis [156] and PARP-mediated necrosis [157–161]. Necroptosis/necrosis leads to the release of intracellular molecules that can promote inflammation via interactions with TLRs and other receptors on immune cells [162, 163]. The postischemic organ also releases proinflammatory cytokines, which promotes the production of inflammatory mediators and inflammation at remote organs [163, 164]. Therefore, the damage that occurs promotes the widespread inflammatory response seen in IR injury. Necrostatin-1 (Nec-1), an inhibitor of RIPK1 and necroptosis, has been shown to be protective against IR injury in the heart, brain and kidney [165–169]; however, caspase inhibitors do not appear to offer any protective benefit against kidney IR injury [165]. This supports the notion that necroptosis/necrosis is principally involved in IR injury. It appears that the effects of tissue ischemia and subsequent reperfusion lead to activation of a variety of necrotic pathways resulting in cell death and an inflammatory response. Although not well studied, this phenomenon is likely to be highly relevant to dermatologic surgery, particularly survival of skin grafts and flaps used during reconstructive surgery after Mohs surgery for skin cancer.
Crohn’s disease is an inflammatory bowel disease involving chronic inflammation primarily of the terminal ileum. Individuals with Crohn’s disease often develop bloody diarrhea and crampy abdominal pain. The inflammation is thought to be triggered by an abnormal immune response to the commensal bacteria of the gut possibly as a result of a disruption of the intestinal epithelial barrier allowing the bacteria to translocate across the bowel wall and stimulate TLR signaling [36, 146–149]. Increased epithelial cell death is a hallmark of intestinal inflammation and believed to be a possible mechanism driving Crohn’s disease [147]. Many studies have shown that the intestinal epithelial cells die by necrosis, specifically necroptosis [36, 149]. This necroptotic death of epithelial cells could be induced by TNF-α in vitro and was associated with increased expression of RIPK3, a key mediator of necroptosis [36, 149]. Studies of mice deficient in caspase 8 or FADD observed spontaneous development of epithelial cell necrosis, intestinal inflammation and loss of Paneth cells similar to that seen in Crohn’s disease [36, 149]. Caspase 8 and FADD are known to be important mediators of apoptosis and regulators of necroptosis, normally keeping the necroptotic pathway silent. Treatment with Nec-1 was protective against the cell death and intestinal inflammation in murine models of Crohn’s disease [36, 149]. Therefore, Crohn’s disease appears to be driven by a disrupted intestinal barrier and inappropriately activated immune response leading to necroptosis of intestinal epithelial cells causing a chronic inflammatory condition.
Crohn’s disease is associated with other chronic inflammatory diseases like psoriasis [150, 151]. The increased incidence of Crohn’s disease among psoriatic patients may be related to the changes in cell death seen in these diseases. The decreased apoptosis seen in psoriasis may be linked to the increased necroptosis observed in Crohn’s disease as apoptotic molecules have been implicated in negatively regulating necroptosis. Further studies are needed to investigate the role of necroptosis in psoriasis.
Therapies Associated with Cell Death
While impaired or abnormal cell death can lead to various dermatological diseases, cell death can also be manipulated for therapeutic purposes commonly used in the field of dermatology. These treatments include PDT, ingenol mebutate, and cryosurgery.
Photodynamic therapy (PDT) is a type of phototherapy in which cell death is induced by light after application of a photosensitizing agent. PDT is important in the treatment of many dermatological conditions such as psoriasis, eczema, non-melanoma skin cancer (NMSC) and melanoma [189]. When the photosensitizing agent is exposed to light, ROS formation results which induces cell death [190–192]. Many studies support the idea that PDT triggers necrotic cell death [192, 193]. However, PDT has also been shown to induce multiple modes of cell death including apoptosis, necrosis and autophagic cell death [192]. PDT using UVA and hypericin, a photosensitizing agent and major component of St. John’s Wort, induced apoptosis in unpigmented cells (KC and non-pigmented melanoma cells) and induced necrosis in pigmented cells (MC and pigmented melanoma cells) [194]. It is likely that the presence of melanin and the site of hypericin localization after UVA exposure contributed to the induced mode of cell death. Hypericin disrupted the melanosome membrane in melanocytes and pigmented melanoma cells causing melanin to leak into the cytoplasm and act as an oxidant to induce cell necrosis. Hypericin localized to the mitochondria in unpigmented cells, leading to mitochondrial damage, cytochrome c release and apoptosis. In pigmented cells, hypericin localized to the ER/Golgi which likely led to cell stress and necrosis. The presence or absence of RIPK3 has also been implicated as a determinant of the modality of cell death induced by PDT, suggesting that PDT may induce necroptosis depending on the cellular context [195]. Therefore, PDT induces and manipulates cell death processes to treat various skin diseases.
Ingenol mebutate is the active ingredient in the sap from the Euphorbia peplus plant [196–198] that has been used to treat a variety of skin lesions [199, 200] including warts, AKs [196, 199, 201, 202], and NMSC [203, 204]. Recent clinical trials have demonstrated that two or three applications of ingenol mebutate is effective in clearing AKs and BCCs. The efficacy of the treatment increased in a dose dependent manner [196, 201, 204] with only mild dose dependent dermatological side effects, confirming that short term use of ingenol mebutate is safe when treating a variety of skin lesions.
Ingenol mebutate has been shown to have a dual mode of action in treating skin lesions. First, rapid necrosis of cells in the targeted lesion is induced [198], which leads to the release of proinflammatory cytokines and activation of the immune system. Second, the activated immune system promotes an inflammatory response and neutrophil-mediated, antibody-dependent cellular cytotoxicity [198, 205], which destroy any residual cells from the lesion. This second mode of action is especially important in completely eradicating the abnormal cells as neutrophil deficient mice treated with ingenol mebutate exhibited tumor regrowth after 25 days compared to neutrophil replete mice who did not exhibit tumor regrowth [205]. Therefore, the inflammatory response and neutrophil-mediated, antibody-dependent cellular cytotoxicity are important to prevent tumor regrowth after the initial necrotic cell death, making ingenol mebutate an effective short term treatment for various skin lesions.
Cryosurgery is a therapeutic technique that involves tissue destruction by freezing, typically using liquid nitrogen [206]. It is widely used in dermatology to treat a variety of benign, premalignant, and malignant skin lesions [207–209]. Cryosurgery treats these lesions by ultimately inducing tissue necrosis [210]. Tissue injury is first induced by freezing, causing direct cell injury [210]. Cell freezing with low cooling rates leads to extracellular ice formation which increases the extracellular solute concentration causing osmotic dehydration of the cell and cell shrinkage [211, 212]. Cell shrinkage and the resulting increased intracellular solute concentration damage the plasma membrane and cellular constituents [213]. The extracellular ice crystals that form can also apply mechanical pressure on the weakened cell, contributing to the plasma membrane damage [212, 214]. Another consequence of freezing is intracellular ice formation, seen at higher cooling rates, which disrupts the cell membrane and cellular organelles leading to cell death [211, 215–220]. Thawing allows recrystallization to occur during which ice crystals can fuse and form larger crystals that are more likely to cause damage to the cellular membrane [211, 221–223]. In addition, as the ice melts, the extracellular fluid becomes briefly hypotonic, which drives water into the cell resulting in cell rupture due to the damaged plasma membrane [224]. Therefore, thawing further contributes to the cell death that was first induced by freezing.
In combination with direct cell injury, vascular stasis plays a major role in the tissue destruction after cryosurgery. Freezing completely terminates circulation in the vessels within the treated region of the tissue [223, 231, 232]. After thawing, there is vasodilation and an increase in blood flow followed by increased vascular leakage due to endothelial cell damage [232], edema, platelet aggregation and blood flow cessation, particularly in the microvasculature [233]. The occlusion of blood flow leads to tissue ischemia and subsequent tissue necrosis [224, 234]. Vascular stasis is believed to be the major mechanism of tissue necrosis induced by cryosurgery.
Some studies suggest that further damage occurs after vascular stasis and tissue necrosis due to an immune response. After the tissue undergoes a necrotic cell death, intracellular contents are exposed which trigger an immune response [235, 236] against the targeted tissue [237]. This suggests that an immune response after cryosurgery contributes to the death of cells that were not immediately killed by freezing and thawing. Edema of and around the treated tissue is observed after cryosurgery, providing further evidence of the activation of the immune system and inflammatory response after tissue necrosis [212, 224]. However, this mechanism is still being elucidated and requires further study. Overall, cryosurgery is an effective therapeutic technique for targeted tissue destruction that induces cell death and an inflammatory response to eliminate unwanted tissue.
Conclusion
As evidenced by the dermatological diseases discussed, cell death can play a significant role in the pathogenic symptoms of a variety of disorders and can significantly influence inflammation. Therefore, it appears that necrosis promotes inflammatory responses while apoptosis has anti-inflammatory effects. However, SJS-TEN spectrum, SLE and GVHD exhibited an increase in apoptosis and an increased inflammatory response, which seems contradictory to the findings that apoptosis is anti-inflammatory/immunosuppressive. These contradictions may be explained by additional factors related to the pathogenesis of these diseases. SJS-TEN spectrum, as recent studies have suggested, may involve necroptosis, which would better explain the increased inflammatory response. SLE not only involves increased apoptosis but also decreased clearance of apoptotic cells which leads to secondary necrosis and drives the inflammatory response/state seen in individuals with SLE. GVHD, like SJS-TEN spectrum and SLE, exhibits an increase in apoptosis and inflammatory response; yet, further studies are needed to explain this discrepancy by looking for necrotic/necroptotic markers in patients with GVHD. Overall, changes in cell death significantly influence inflammation in a variety of dermatological diseases. In particular, this inflammatory response seems to be related to a decrease in apoptosis and/or an increase in necrosis. Understanding this concept has helped and will continue to help develop more advanced techniques to treat these inflammatory dermatological diseases.
Questions
- 1.
How can a keratinocyte undergoing apoptotic cell death be differentiated from one that is dying by necrosis/necroptosis?
- A.
DNA “laddering”
- B.
Cellular “blebbing” on electron microscopy
- C.
Lack of phosphorylation of MLKL
- D.
Cleavage of RIP1
- E.
All of the above
- F.
None of the above
- A.
Correct Answer: (E) All of the above cellular and biochemical events described above are hallmarks of apoptosis, and distinguish this form of cell death from necroptosis
- 2.
What type of cell death may be advantageous to promote in a patient presenting with symptoms of hyper-immune cell activation and several areas of inflammatory skin lesions?
- A.
Necroptosis
- B.
Pyroroptosis
- C.
Apoptosis
- D.
Autophagy
- A.
Correct Answer: (C) Apoptosis is a non-inflammatory type of cell death, and can actually suppress antigen presenting cell function, and thus dampen immune cell activation. This is not true for the other types of cell death or autophagy
- 3.
Get Clinical Tree app for offline access
Crohn’s disease is associated most strongly with which mode of cell death?
- A.
ApoptosisRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



- A.