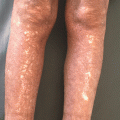
Fig. 16.1
S. aureus cellulitis on the lower leg. The involved skin characteristically shows signs of inflammation, including erythema, warmth, edema, and pain (Photograph is courtesy of the Victor D. Newcomer collection at UCLA and Logical Images, Inc.)
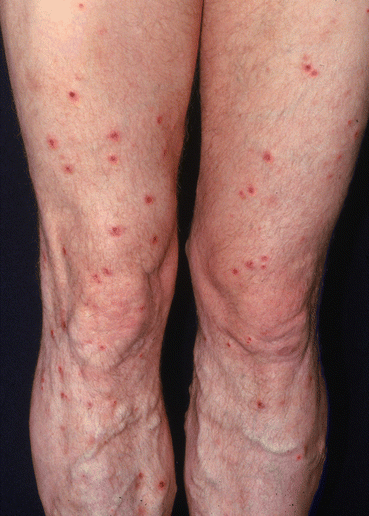
Fig. 16.2
S. aureus folliculitis/furuncles (boils). Typically, infected hair follicles present as follicularly-based erythematous, warm, edematous, and pus-filled papules and nodules (Photograph is courtesy of the Victor D. Newcomer collection at UCLA and Logical Images, Inc.)
Innate Immune Responses Against S. aureus
Innate immune responses are directed against conserved components of microorganisms called pathogen associated molecular patterns (PAMPs) [18, 19]. The host cellular receptors that recognize different PAMPs are called pattern recognition receptors (PRRs) [18, 19]. PAMPs are predominantly expressed by microorganisms and not by host cells, thus enabling the host’s immune system to recognize the pathogen rather than self [18, 19]. In this section, soluble factors of the innate immune system such as antimicrobial peptides and complement will be discussed, followed by a review of the known PRRs, inflammasomes and the role of innate immune system cells such as neutrophils and monocytes/macrophages in host defense against S. aureus.
Soluble Mediators of Innate Immunity Against S. aureus
Antimicrobial Peptides
Antimicrobial peptides are polypeptides that have antimicrobial activity at physiologic conditions and are believed to function by disrupting bacterial membranes [20–23]. There are several human antimicrobial peptides involved in skin host defense, including defensins (α and β), cathelicidin, RNase 7, dermcidin and REG3A (Table 16.1) [20–24]. Alpha-defensins (also called human neutrophil peptides, HNPs) are produced by neutrophils, whereas β-defensins are produced predominantly by epithelial cells (including keratinocytes) and are also produced by macrophages and dendritic cells [25]. Cathelicidin is produced constitutively by neutrophils and can be induced in epithelial cells, including keratinocytes [20, 26, 27].
Table 16.1
Soluble mediators and pattern recognition receptors that contribute to host defense against S. aureus skin infections and colonization
Cellular expression in skin | S. aureus evasion mechanisms | Mechanisms of action | |
|---|---|---|---|
Antimicrobial peptides | |||
α-defensins (human neutrophil peptides [HNPs]) | Neutrophils | Staphylokinase, MprF, dltABCD operon | Antimicrobial activity, chemotaxis of T cells and immature dendritic cells |
hBD2 | Keratinocytes macrophages DCs | IsdA, dltABCD operon | Antimicrobial activity, chemotaxis of immature dendritic cells and memory CD4+ T cells via CCR6 |
hBD3 | Keratinocytes | dltABCD operon | |
LL-37 | Keratinocytes macrophages neutrophils | IsdA, aureolysin, MprF, dltABCD operon | Antimicrobial activity, chemotaxis of neutrophils, monocytes and T cells via FPRL1 |
Dermcidin | Eccrine glands | Extracellular proteases, dltABCD operon | Antimicrobial activity |
RNase 7 | Keratinocytes | dltABCD operon | Antimicrobial activity |
REG3A | Keratinocytes | Antimicrobial activity | |
Complement | |||
C3a, C5a | Serum | SCIN | Chemotaxis of neutrophils |
C3b | Serum | SCIN, Efb, C4BP, staphylokinase | Opsonophagocytosis |
Mannose-binding lectin (MBL) | Serum | SCIN, Efb, C4BP, staphylokinase | Activation of the lectin complement pathway, opsonophagocytosis, |
Toll-like receptors (TLRs) | |||
TLR2 | Multiple | SSL3 | Recognize S. aureus lipopeptides, LTA, PGN |
TLR9 | Multiple | Recognize S. aureus DNA in endosomes | |
NOD2 | Multiple | Recognize cytosolic muramyl-dipeptide (a breakdown product of S. aureus PGN) | |
Inflammasomes | |||
NLRP3/ASC | Multiple | Activated by S. aureus toxins, ATP & K+ efflux to induce pro-IL-1β processing | |
AIM2/ASC | Multiple | Activated by cytosolic S. aureus DNA to induce pro-IL-1β processing | |
Formyl peptide receptors | |||
FPR1, FPR2, FPR3 | Macrophages Neutrophils | CHIPS | Recognize formylated peptides to promote chemotaxis, phagocytosis and oxidative burst |
Tumor necrosis factor-α receptor 1 (TNFR1) | |||
TNFR1 | Multiple | BINDS S. aureus protein A to promote inflammation | |
Peptidoglycan recognition proteins (PGLYRP1) | |||
PGLYRP1 | Neutrophils | BINDS S. aureus PGN to induce antimicrobial activity | |
PGLYRP2 | Keratinocytes | BINDS S. aureus PGN | |
PGLYRP3,4 | Keratinocytes Hair follicles Sebaceous glands Sweat glands | BINDS S. aureus PGN | |
There are six known human α-defensins (HNP1-6), which constitute ~50 % of the peptides found within neutrophil granules [28]. Notably, HNPs 2 and 5 (and not HNPs 1, 3, 4, and 6) have antimicrobial activity against S. aureus in vitro [29]. The antimicrobial activity of α-defensins is likely important in host defense since S. aureus produces proteins such as staphylokinase, which directly binds to α-defensins to inhibit their activity [30]. In addition, MprF and products of the dltABCD operon reduce the negative charge of the bacterial membrane via lysinylating phosphatidylgycerol [31] and alanylating teichoic acids [32] in the bacterial cell envelope, respectively, to reduce the activity of α-defensins.
There are four well-characterized human β-defensins (HBD1-4), which are expressed by various epithelial cells, including keratinocytes, as well as by activated monocytes/macrophages and dendritic cells. HBDs 1, 2, and 4 have been shown to have only a weak bacteriostatic effect against S. aureus in vitro [33, 34]. In contrast, HBD3 has strong in vitro bactericidal activity against S. aureus [35]. hCAP-18 is the only known member of the human cathelicidin family and is the precursor to the active cleaved C-terminal peptide LL-37 [36–38]. Like HBD3, LL-37 has been shown to have potent antimicrobial activity against S. aureus [36–38].
Keratinocyte production of HBD2, HBD3, and LL-37 can be induced by live or heat-killed S. aureus and by S. aureus components, including lipopeptides and lipoteichoic acid (LTA) via activation of TLR2 [39–42]. Thus, human keratinocytes can upregulate the production of HBD2 and HBD3 in response to S. aureus or its components, thereby increasing the innate immune response in the skin. Interestingly, activation of the epidermal growth factor receptor (EGF-R) by wounding of human skin in vitro resulted in increased production of HBD3 and antimicrobial activity against S. aureus [43, 44]. Thus, wounded skin may resist infection by S. aureus via production of HBD3 [43, 44]. In addition, vitamin D has been shown to increase LL-37 production by keratinocytes, neutrophils and monocytes/macrophages [45–47], suggesting vitamin D may also promote host defense against S. aureus [48, 49]. Of note, the important role of HBDs and LL-37 in host defense against S. aureus in skin is illustrated by the expression of the surface protein, iron surface determinant A (IsdA), by S. aureus, which renders the bacteria resistant to hBD2 and LL-37 by decreasing the bacterial membrane hydrophobicity [50]. S. aureus also produces aureolysin, a protease that cleaves LL-37 to inactivate its activity [51]. Moreover, MprF is not only effective against α-defensins (see above) but can also neutralize the activity of LL-37 [52].
Newer antimicrobial peptides have been identified in skin that have activity against S. aureus, including RNase 7, which is produced by keratinocytes and can prevent skin colonization with S. aureus [53], dermcidin, which is produced by eccrine glands and secreted in human sweat [54, 55], and REG3A [24], which is produced by keratinocytes and the mouse homolog, REG3γ, has activity against S. aureus pneumonia [56]. Of note, S. aureus secretes extracellular proteases that degrade and neutralize the activity of dermcidin [57]. It should be highlighted that the S. aureus-derived products of the dltABCD operon appear to have a global effect on inhibiting antimicrobial peptide activity by decreasing the negative charge on the bacterial surface, since a mutant S. aureus strain deficient in D-alanylated teichoic acids (dltA mutant) was more susceptible to killing by HBD2, HBD3, LL-37, RNase 7 and dermcidin [58].
Antimicrobial peptides may also be involved in the pathogenesis of certain inflammatory skin diseases. HBD2 and HBD3 are expressed at increased levels in the hyperproliferative and inflammatory skin disease, psoriasis, which has been associated with a Th1 and Th17 cytokine profile [33, 35, 59, 60]. In contrast, levels of HBD2, HBD3, and LL-37 are expressed at significantly lower levels in atopic dermatitis, another inflammatory skin disease that is associated with a Th2 cytokine profile [59, 60]. These findings provide an explanation of why S. aureus colonization and superinfection is more frequently seen in atopic dermatitis and rarely in psoriasis.
Lastly, antimicrobial peptides not only have microbicidal activity, but also promote the recruitment of immune system cells to the site of infection. For example, HNPs promote chemotaxis of T cells and immature dendritic cells, HBDs promote chemotaxis of immature dendritic cells and memory CD4 T cells, and LL-37 promotes chemotaxis of neutrophils, monocytes, and T cells [61–63]. The chemotactic activity of HBDs and LL-37 is mediated by the chemokine receptor CCR6 and the formyl peptide receptor-like 1 (FPRL1), respectively [61–63].
Complement
The complement system includes a family of serum proteins, proteolytic fragments and cell surface receptors [64, 65]. There are three main functions of complement activation: (1) direct killing of bacteria via formation of the membrane attack complex (MAC), which perforates cell membranes; (2) promotion of phagocytosis by opsonizing the bacterial surface with complement components such as C3b; and (3) recruitment of immune system cells to the site of infection by generating the complement chemoattractant peptides C3a and C5a [64, 65]. There are three pathways of complement activation: the classical, alternative and lectin pathways [64, 65]. Each pathway requires generation of an enzyme complex called the C3 convertase [64, 65]. In the classical pathway, generation of the C3 convertase involves the interaction of complement components with natural IgM antibody or antigen specific IgG antibody that is bound to antigens of the pathogen [64, 65]. In the alternative pathway, there are low levels of direct activation of the C3 convertase by components of the pathogen [64, 65]. In the lectin pathway, the C3 convertase is activated via recognition of carbohydrate groups on the surface of the bacteria via mannose-binding lectin (MBL) or via H-, L-, or M-ficolin and MBL-associated serine proteases (MASP1, 2 and 3) [66].
There have been several reports demonstrating the key role of complement in host defense against S. aureus (Table 16.1). First, in a mouse model of S. aureus-induced arthritis and bacteremia, depletion of complement components resulted in higher mortality and significantly decreased neutrophil recruitment and impairment of phagocytosis [67]. In another study, complement mediated opsonization by C3b as well as activation of complement receptor 1 (CD35) (the primary receptor for C3b) were more critical in controlling S. aureus bacteremia than C5a or generation of the MAC [68].
Table 16.2
Soluble mediators and pattern recognition receptors that contribute to host defense against group A Streptococcus skin infections
Cellular expression in skin | Group A Streptococcus evasion mechanisms | Mechanisms of action | |
|---|---|---|---|
Antimicrobial peptides | |||
α-defensins (human neutrophil peptides [HNPs]) | Neutrophils | SIC, generation of dermatan sulphate, dltABCD | Antimicrobial activity, chemotaxis of T cells and immature dendritic cells |
hBD2 | Keratinocytes, macrophages, DCs | SIC, dltABCD operon | Antimicrobial activity, chemotaxis of immature dendritic cells and memory CD4+ T cells via CCR6 |
hBD3 | Keratinocytes | SIC, DRS, dltABCD operon | |
LL-37 | Keratinocytes, macrophages neutrophils | SIC, SpeB, dltABCD operon | Antimicrobial activity, chemotaxis of neutrophils, monocytes and T cells via FPRL1 |
Complement | Serum | M-protein | Complement cascade |
C5a | Serum | C5a peptidase (ScpA) | Chemotaxis of neutrophils |
C5b-C7 | Serum | SIC | MAC (membrane attack complex) |
Toll-like receptors (TLRs) | |||
TLR9 | Multiple | Sda1 | Recognize group A Streptococcus DNA |
NOD2 | Multiple | Recognize cytosolic muramyl-dipeptide (a breakdown product of group A Streptococcus PGN) | |
Inflammasomes | |||
NLRP3/ASC inflammasome | Multiple | Activated by group A Streptococcus streptolysin O to induce pro-IL-1β processing | |
MBL is also important in host defense against S. aureus infections [69–71]. For example, individuals with an MBL gene mutation, who have impaired MBL-dependent opsonization, suffer from recurrent S. aureus infections [72]. MBL as well as IgG directed against glycoepitopes on teichoic acid facilitates complement-mediated opsonophagocytosis of S. aureus [73, 74]. In addition, MBL-deficient mice had only a slightly decreased survival whereas C3-deficient mice and mice deficient in both MBL and C3 had markedly decreased survival compared with wildtype mice after an intravenous challenge of S. aureus [69, 70]. Thus, C3 and complement activation may play a more important role than MBL in host defense against S. aureus [69, 70]. Finally, MBL can bind to S. aureus in vitro, resulting in increased phagocytosis [71]. Taken together, MBL is involved in activation of the lectin complement pathway and in opsonization of S. aureus.
S. aureus has several mechanisms to counteract complement activity. S. aureus produces a protein called staphylococcus complement inhibitor (SCIN), which blocks activation of the complement cascade by inhibiting the C3 convertase [75]. In addition, S. aureus secretes extracellular fibrinogen-binding protein (Efb) and extracellular complement-binding (Ecb) proteins, which bind C3 and blocks C3 opsonization [76–78]. S. aureus also produces C4b-binding protein (C4BP) that cleaves CD4b into inactive forms iC4b and C4d, which decreases C3b-mediated opsonization [79]. Lastly, staphylokinase (SAK) inhibits opsonization of S. aureus and subsequent phagocytosis by converting plasminogen into plasmin on the bacterial surface, which leads to removal of the anti-staphylococcal opsonins IgG and C3b [80].
Pattern Recognition Receptors that Recognize Components of S. aureus
Toll-Like Receptors
Toll-like receptors (TLRs) are important PRRs involved in host defense against a variety of pathogenic microorganisms, including S. aureus [18, 19]. Activation of TLRs initiates several signaling cascades including NF-kB activation, ultimately leading to production of cytokines, chemokines, antimicrobial peptides and up-regulation of costimulatory and adhesion molecules involved in innate and adaptive immune responses [18, 19].
Of the known human TLRs (1-10), TLR2 has been the most implicated in host defense against S. aureus (Fig. 16.3, Table 16.1). TLR2 is expressed on the cell surface of numerous cell types in the skin, including keratinocytes, Langerhans cells, monocytes/macrophages, dendritic cells, mast cells, endothelial cells, fibroblasts and adipocytes [81–89]. TLR2 can be activated by live or heat-killed S. aureus as well as the S. aureus components, lipopeptides, lipoteichoic acid (LTA) and peptidoglycan (PGN) [90].
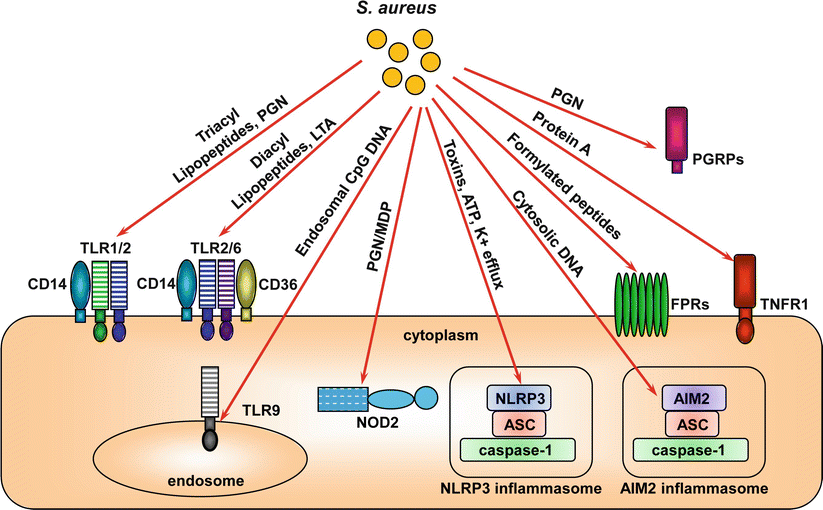
Fig. 16.3
Pattern recognition receptors (PRRs) of host cells involved in recognizing components of S. aureus and initiating immune responses. The S. aureus components recognized by these PRRs and the cellular localization of these PRRs are shown
With regard to S. aureus skin infections, TLR2-deficient mice develop larger skin lesions than wildtype mice after S. aureus skin inoculation [91–93]. Human keratinocytes also express TLR2 and can be activated by live or heat-killed S. aureus and S. aureus components, resulting in increased production of cytokines such as IL-1β, IL-8, TNFα and production of HBD2 and HBD3 [82, 85, 86, 94]. Polymorphisms in TLR2 have been linked to increased severity of atopic dermatitis [95–98], which is frequently associated with superinfection by S. aureus [99, 100]. In addition, TLR2 was found to enhance barrier repair in human skin and the reduced expression of TLR2 in atopic dermatitis may contribute to the impaired skin barrier in this disease [101]. Most recently, a study in mice found that activation of TLR2 promoted the shift from acute Th2-mediated dermatitis to chronic cutaneous inflammation in a mechanism that involved IL-4 suppression of IL-10 [102]. This study provides additional rationale for the therapeutic targeting of IL-4, which is currently under investigation against atopic dermatitis in human trials [103].
Since S. aureus lipopeptides, LTA and PGN have distinct biochemical structures, it was unclear how one receptor could recognize such a broad spectrum of molecules [104]. However, TLR2 interacts with other TLRs and additional co-receptors, which enables TLR2 to recognize these different ligands. TLR2 heterodimerizes with TLR1 or TLR6 to recognize tri-acyl and di-acyl lipopeptides, respectively [105, 106]. Therefore, the ability of the host to recognize certain lipopeptides depends on the formation of TLR2 heterodimers. CD14, a membrane protein that lacks an intracellular signaling domain, was initially characterized as a TLR4 co-receptor for LPS of Gram-negative bacteria [104]. However, CD14 also acts as a TLR2 co-receptor to recognize S. aureus LTA and PGN [107, 108]. CD14 is likely involved in cutaneous immunity against S. aureus since studies have demonstrated that increased CD14 expression in keratinocytes inhibited growth of S. aureus [49, 109]. In addition, CD36 represents another TLR2 co-receptor involved in the recognition of S. aureus LTA (which is di-acylated) and in the activation of signaling via the TLR2/6 heterodimer [92, 110]. The importance of TLR2 is exemplified by the existence of superantigen-like protein 3 (SSL3), a S. aureus-derived factor that binds and inhibits the function of TLR2 [111, 112].
TLR9 is an intracellular TLR that is found spanning the membranes of endosomes that has been shown to recognize hypomethylated CpG (cytosine-phosphate-guanosine) motifs of bacterial DNA and is involved in promoting type I interferon responses [113–115]. Although the role of TLR9 against S. aureus skin infections is not entirely clear, TLR9-deficient mice have been found to have enhanced clearance against a S. aureus pneumonia infection, suggesting that TLR9 responses may not be associated with protection against S. aureus infections [116].
Nucleotide-Binding Oligomerization Domain Proteins (NOD1 and NOD2)
In contrast to TLR2, nucleotide-binding oligomerization domain proteins (NOD1 and NOD2) are found in the cytosol and detect breakdown products of PGN [117]. NOD1 recognizes breakdown products of Gram-negative PGN [117]. In contrast, NOD2 recognizes muramyl dipeptide (MDP), which is a breakdown product of both Gram-positive and Gram-negative PGN [117], and has been shown to recognize MDP-derived from S. aureus PGN (Fig. 16.3, Table 16.1) [118]. After ligand detection, NODs activate a signaling pathway that results in NF-kB activation and transcription of host genes involved in innate and acquired immune responses [117].
Since NOD2 is a cytoplasmic receptor, this calls into question whether an intracellular PRR could be involved in recognition of a S. aureus infection, since S. aureus has classically been considered an extracellular pathogen. However, several studies have found that S. aureus can invade the cytoplasm of various cells, including keratinocytes, epithelial cells, fibroblasts, endothelial cells, osteoblasts and neutrophils [119]. Once S. aureus enters the cytoplasm, host and/or bacterial enzymes may break down S. aureus PGN into MDP that can be recognized by NOD2 [119]. A few studies have found that NOD2 is involved in host defense against S. aureus skin infections in mice [91, 120]. Moreover, NOD2 expressed by keratinocytes can induce inflammatory cytokines such as IL-1β, IL-6 and IL-17C [121, 122], which promote antimicrobial mechanisms (e.g., neutrophil recruitment and activation and expression of antimicrobial peptides) against S. aureus [91, 120, 122]. Finally, NOD2 may also play a role against human S. aureus infections because polymorphisms in NOD2 (like TLR2) were identified in patients with atopic dermatitis [123, 124].
Inflammasomes
IL-1β plays a critical role in the recruitment of neutrophils to the site of S. aureus infection in the skin [91, 93, 125, 126]. Although, PRRs such as TLRs and NODs can induce transcription and translation of pro-IL-1β (as well as pro-IL-18) [127], a second signal is required to induce cleavage of pro-IL-1β (and pro-IL-18) into its active and secreted form. This process is typically mediated by inflammasomes, which are cytoplasmic protein complexes that regulate caspase-1-mediated proteolytic processing of pro-IL-1β into active IL-1β (Fig. 16.3, Table 16.1) [128–130]. There are several known inflammasome complexes, which are reviewed elsewhere [128–130]. However, the NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome has been the primary inflammasome complex implicated in IL-1β processing in the context of S. aureus infections (Fig. 16.3). In S. aureus-infected macrophages in vitro, the NLRP3-inflammasome processes IL-1β in a mechanism that involves activation of this inflammasome by S. aureus toxins (α-, β-, and γ-toxins and Panton-Valentine leukocidin [PVL]), ATP/P2X7R activation and phagosomal rupture [131–136]. This may be relevant to S. aureus skin infections in vivo, since mice deficient in ASC (apoptosis-associated speck-like protein containing a CARD), which is a critical component of the NLRP3 inflammasome, has the same impaired immune response against S. aureus skin infections as IL-1β-deficient mice [126]. In addition, the AIM2 (absent in melanoma 2) inflammasome, which like NLRP3 requires interaction with ASC [137–139], was recently shown to be important for host defense against a S. aureus brain abscess infection [140]. AIM2 recognizes cytosolic bacterial DNA [137–139] and the AIM2/ASC inflammasome may recognize S. aureus DNA in the cytosol to promote host defense. Interestingly, in certain cell types, such as neutrophils, processing of IL-1β can be mediated in an inflammasome-independent manner via the activity of other proteases, such as elastase, proteinase 3, cathepsins and matrix metalloproteinases (MMPs) [141–145]. In addition, neutrophils have been shown to be an important source of IL-1β during S. aureus skin infections in mice and it is likely that both inflammasome and non-inflammasome mechanisms contribute to IL-1β processing and release in neutrophils [91].
Formyl Peptide Receptors (FPRs)
Bacteria but not mammalian cells produce peptides and proteins containing formylated methionine [146]. These formylated non-self peptides and proteins can be recognized by formyl peptide receptors (FPRs) on host cells (including macrophages and neutrophils) (Fig. 16.3, Table 16.1) [146]. There are three known human FPRs, FPR1, FPR2 and FPR3 [146]. In particular, human FPR1 and the mouse ortholog mFPR1 as well as human FPR2 have been implicated in promoting neutrophil chemotaxis, phagocytosis and oxidative burst against S. aureus infections [147–149]. The importance of FPRs in host defense against S. aureus is demonstrated by the activity of the chemotaxis inhibitory protein of staphylococci (CHIPS), which blocks the chemotactic activity of FPR1, FPR2 and FPR-like 1 inhibitory proteins (FLIPr and FLIPr-like) [150, 151].
Tumor Necrosis Factor-α Receptor 1 (TNFR1)
TNF-α receptor 1 (TNFR1) is a receptor for TNF-α that is expressed on many different cell types. S. aureus protein A, which is known to bind the Fc portion of antibody, was found to activate TNFR1 to trigger production of proinflammatory cytokines and chemokines and contributed to virulence in a mouse model of S. aureus pneumonia (Fig. 16.3, Table 16.1) [152, 153]. A similar mechanism of protein A and TNFR1 interaction has been shown to occur in human keratinocytes, which induces production of proinflammatory cytokines and chemokines [154]. However the relevance of TNFR1 in contributing to the pathogenesis of S. aureus skin infections in vivo is unclear since TNFR1-deficient mice had similar lesion sizes and bacteria clearance as wildtype mice during an in vivo S. aureus skin infection [93].
Peptidoglycan Recognition Proteins (PGRPs)
In humans, there are four PGRP genes (PGLYRP1, 2, 3, 4, formally named PGRP-S, -L, -Iα, and -Iβ based on their short [S], long [L] and intermediate [I] transcript lengths) [155]. All of these gene products are secreted proteins (Fig. 16.3, Table 16.1) [155]. PGLYRP1 is expressed within tertiary granules of neutrophils and it has been shown to bind S. aureus PGN to promote antimicrobial activity [156, 157]. PGLYRP2 is expressed in keratinocytes and has an active amidase that cleaves S. aureus PGN [158]. PGLYRP3 and PGLYRP4 are also expressed in the kertainocytes as well hair follicles, sebaceous glands and sweat glands [159]. It is unclear whether these PGRPs play an important host defense role against S. aureus skin infections. However, PGLYRP2-deficient mice had no immune impairment against a systemic challenge with S. aureus [160].
Cellular Innate Immune Responses Against S. aureus
Neutrophils
Neutrophils are first responders of the innate immune system and are recruited to sites of S. aureus infection [15, 16]. Neutrophilic responses are thought to be crucial for immunity against both primary and recurrent S. aureus infections since patients with congenital or acquired defects in neutrophil number, recruitment or function (e.g., congenital conditions such as chronic granulomatous disease and acquired conditions such as neutropenic chemotherapy patients or patients with impaired neutrophil function in diabetes or renal insufficiency) are highly susceptible to S. aureus infections in many tissues and organs, including the skin [127]. Keratinocytes and other resident skin cells produce neutrophil-attracting chemokines such as neutrophil chemotactic factor IL-8 (CXCL8), growth-related oncogene-α, -β, -γ (GRO-α, -β, -γ), neutrophil-activating peptide-2 (NAP-2; CXCL7) and epithelial cell-derived neutrophil-activating peptide-78 (ENA-78, CXCL5) [161]. All of these chemokines contain glutamic acid-leucine-arginine (ELR) residues preceding the first cysteine and activate the receptors CXCR1 and CXCR2 on neutrophils to promote chemotaxis and are thus called ELR+ chemokines [161]. The antimicrobial peptide LL-37 and the complement components C3a and C5a are also strong neutrophil chemoattractants [161, 162]. In addition, neutrophils themselves release leukotrienes, which are proinflammatory molecules that are chemoattractant for most leukocytes [161].
One of the main neutrophil functions is to engulf microbes into a phagosome, which fuses with a lysosome to form a phagolysosome (Fig. 16.4) [15, 16]. In the phagosome, reactive oxygen species (ROS) are produced such as superoxide (O2−), hydrogen peroxide (H2O2), and hyperchlorous acid (HOCl) by the enzymes NADPH oxidase, superoxide dismutase and myeloperoxidase (MPO), respectively. These ROS are toxic to certain bacterial pathogens, but S. aureus is somewhat resistant to ROS-mediated killing alone [15, 163]. However, ROS also promote killing of bacteria such as S. aureus by producing a charge across the phagocytic vacuole membrane, resulting in K+ influx and release of proteases and antimicrobial peptides from neutrophil granules into the vacuole [16]. Some of the components of neutrophil granules that are important in bacterial killing include proteinases (e.g. cathepsin G, elastase, and proteinase 3), α-defensins, lysozyme, acid hydrolases, lactoferrin (which sequesters iron and copper), transcobalamin II (which binds cyanocobalamin [vitamin B12]), and neutrophil gelatinase-associated lipocalin (NGAL) [164]. NGAL is an antimicrobial protein that binds to bacterial siderophores and blocks their ability to extract iron needed for bacterial growth [164]. If neutrophils escape the phagosome and enter the cytoplasm, approximately 40–50 % of the protein found in the cytoplasm is comprised of calprotection, a complex of S100A8 and S100A9 proteins, which chelates Mn2+ and Zn2+ and sequesters these essential nutrients to prevent bacterial growth [164]. Finally, neutrophils can release antimicrobial peptides, proteases and chromatin through NETs (neutrophil extracellular traps), which trap and promote antimicrobial activity against S. aureus [165, 166].
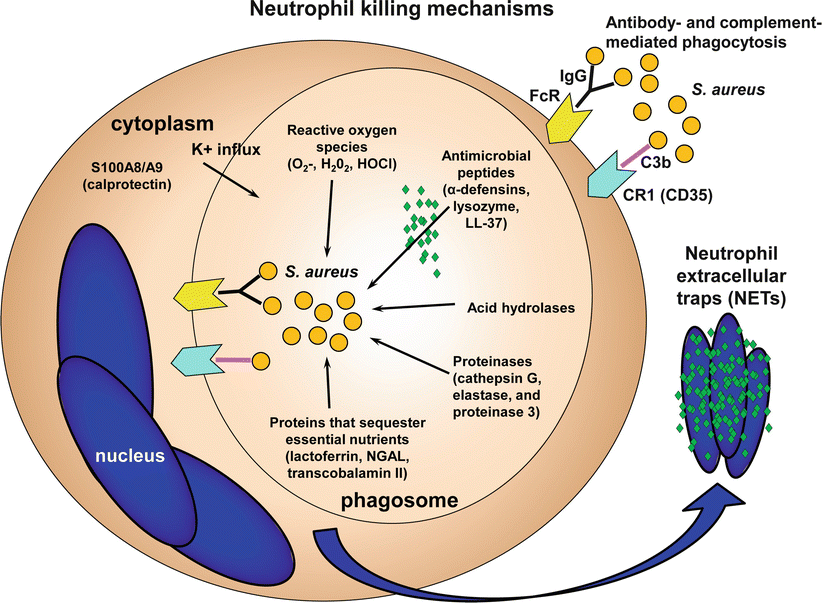
Fig. 16.4
Neutrophil antimicrobial mechanisms against S. aureus
Recently, a host defense circuit has been uncovered that triggers neutrophil recruitment to a site of S. aureus skin infection in mice (Fig. 16.5). This process begins with recognition of the S. aureus skin infection by PRRs such as TLR2, NOD2 and FPRs, which leads to production and secretion of IL-1β in a mechanism involving activation of the NLRP3/ASC inflammasome [91, 93, 126]. IL-1β then acts on IL-1R-expressing resident skin cells and subsequent signaling via MyD88 leads to production of neutrophil-attracting chemokines (KC, MIP2) and granulopoiesis factors (G-CSF and GM-CSF) to mediate neutrophil recruitment [91, 93, 126]. Interestingly, a major cellular source of IL-1β at the site of S. aureus infection in the skin were neutrophils, which amplified and sustained the neutrophilic response for optimal abscess formation and bacterial clearance [91]. It should also be mentioned that IL-1α also contributed to IL-1R/MyD88-mediated host defense against a superficial S. aureus skin infection in mice [125], which is consistent with the identification of an IL-1α autocrine signaling loop in keratinocytes to continuously produce neutrophil-attracting chemokines [167, 168]. A similar mechanism involving IL-1α/β in host defense may exist in humans since pediatric patients with a deficiency in MyD88 or IRAK4 (signaling molecules downstream of IL-1Rs and TLRs) are highly susceptible to pyogenic bacterial infections, including S. pneumoniae, S. aureus and P. aeruginosa [169–171]. In these patients, the S. aureus infections were limited to the skin in most cases (84 %) [169–171], which is different than patients with impaired neutrophilic responses because they have a systemic susceptibility to S. aureus infections [127]. Furthermore, the MyD88/IRAK4-deficient patients developed pus at sites of infection in the absence of neutrophilia, suggesting an impaired ability to sustain the neutrophilic response [170], which is consistent with the findings in mice [91, 93, 126].
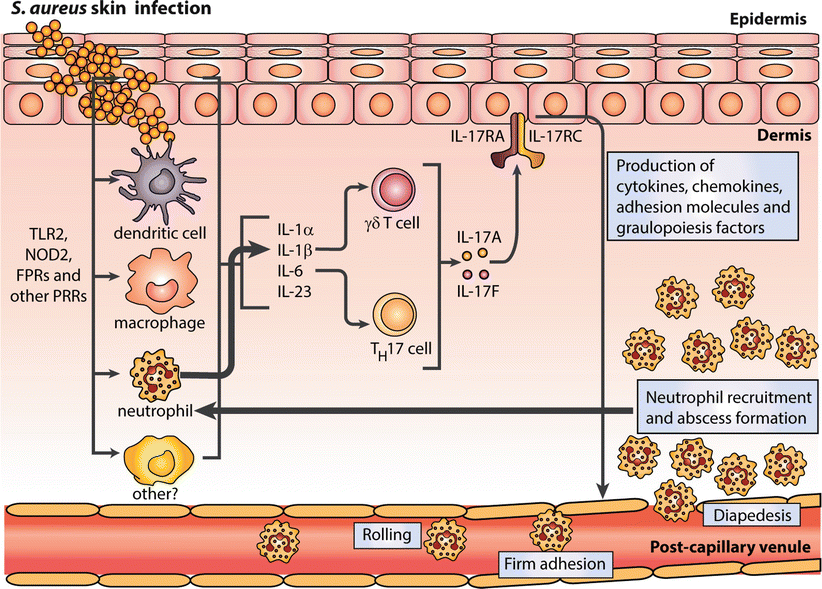
Fig. 16.5
Proposed host defense circuit for neutrophil recruitment to a site of S. aureus infection in the skin. Recognition of S. aureus components by PRRs (TLR2, NOD2, FPR1) and activation of cells in the skin to produce IL-1α, IL-1β, IL-6 and IL-23, which are required to activate γδ T cells and/or Th17 cells to produce IL-17A and IL-17 F. IL-17A/F subsequently activates IL-17 receptors, expressed primarily on keratinocytes, to produce cytokines, chemokines, adhesion molecules and granulopoiesis factor to promote neutrophil recruitment from the circulation to the site of infection. Finally, neutrophils, themselves, are important sources of IL-1β, which can amplify and sustain the neutrophil recruitment response for optimal abscess formation
The critical role of neutrophils in host defense against S. aureus is further illustrated by the existence of mechanisms that S. aureus possesses to inhibit neutrophil recruitment and function [172]. For example, S. aureus inhibits neutrophil extravasation by producing staphylococcal superantigen-like 5 (SSL5) that blocks P-selectin-mediated neutrophil rolling, and extracellular adherence protein (Eap) that binds to ICAM-1 to inhibit adherence to endothelium [76]. S. aureus also inhibits neutrophil chemotaxis via production of Staphopain A, which blocks activity of CXCR2 chemokines [173], and CHIPS that inhibits neutrophil chemotaxis by blocking CD5aR, FPR1, FPR2, FPRL1 and FLIPr and FLIPr-like receptors [150, 151]. S. aureus also produces factors that inhibit neutrophil function. For example, the yellow carotenoid pigment of S. aureus, staphyloxanthin, which is responsible for its golden color, is an antioxidant that blocks ROS-mediated killing of S. aureus [174]. In addition, S. aureus produces catalase and alkyl hydroperoxide reductase, which converts hydrogen peroxide to water, and two superoxide dismutase enzymes, which degrade superoxide, to impair ROS-mediated killing [172]. S. aureus also produces nuclease and adenosine synthase that degrade NETs, thereby evading their antimicrobial function [175, 176].
Monocytes/Macrophages
Similar to neutrophils, monocytes/macrophages are recruited to the site of a S. aureus infection and are important in phagocytosing S. aureus. Monocytes/macrophages (as well as neutrophils) express Fc and complement receptors that facilitate phagocytosis by recognizing immunoglobulin or complement components opsonized on the bacterial surface [172, 177]. The importance of phagocytosis is exemplified by the existence of several mechanisms that S. aureus utilizes to evade this process [177]. For example, S. aureus has protein A on its surface that binds the Fc portion of IgG, resulting in the binding of IgG in an incorrect orientation for detection by Fc receptors [177]. In addition, fibrinogen binding proteins and clumping factor A (ClfA) bind fibrinogen and impair macrophage phagocytosis [177]. S. aureus also secretes toxins that are pore-forming proteins that damage membranes of host cells such as macrophages leading to lysis and the prevention of phagocytosis [172]. There are two main families of these pore-forming toxins: (1) single-component α-hemolysin or α-toxin and (2) biocomponent leukotoxins, including γ-hemolysin or γ-toxin, Panton-Valentine leukocidin (PVL), leukocidin A/B (LukAB), LukED, LukGH, and leukocidin M/F-PV-like [172]. The toxins facilitate lysis of host cells by interacting with specific targets, some of which have been identified. Alpha-toxin targets ADAM10 [178], PVL targets C5a receptors [179], LukAB targets CD11b [180] and LukED targets CCR5, CXCR1 and CXCR2 [181, 182]. Finally, S. aureus possesses phenol soluble modulins, including four PSMα peptides (PSMα1-PSM α4), PSMβ1, PSMβ2, and PSMδ (δ-toxin), which have the ability to lyse human erythrocytes and leukocytes, including neutrophils [183]. In addition, CA-MRSA strains are known to produce high levels of PSMα peptides that contribute to the enhanced virulence of USA300 isolates [184].
Adaptive Immune Responses Against S. aureus
The innate immune system provides the first line of defense against microbial pathogens, while the cell-mediated and humoral immune responses of adaptive immunity are later recruited. The adaptive immune system can be divided into T cell- and B cell-mediated immune responses and the role of these adaptive immune responses against S. aureus will be discussed in this section.
T Cell Immune Response
A number of different observations have provided evidence that T cells play an important role in host defense against S. aureus skin infections. First, patients with HIV infection are at an increased risk for colonization and skin infection with S. aureus [185–188]. In addition, the low serum CD4+ T cell counts of HIV patients is a risk factor for S. aureus infection [185].
Second, patients with the inflammatory skin disease atopic dermatitis, which is associated with a Th2 cytokine profile (i.e. IL-4, IL-13, and IL-10), have increased colonization and superinfection with S. aureus [189, 190]. Although the reason for the increased S. aureus superinfection in atopic dermatitis is likely multifactorial (including a defective epidermal barrier and impaired innate immune responses such as decreased expression of antimicrobial peptides), Th2 cytokines have also been implicated. For example, IL-4 has been shown to increase the expression of fibronectin and fibrinogen receptors on host cells, which promotes more efficient binding of S. aureus to the stratum corneum [191]. In addition, S. aureus via the S. aureus-derived factors fibronectin-binding protein (fnbp) and clumping factor (Clf, also known as fibrinogen-binding protein) more efficiently bind to skin from atopic dermatitis patients [192]. Also in atopic dermatitis, S. aureus produces superantigens such as staphylococcal enterotoxins A and B (SEA and SEB) and toxic shock syndrome toxin-1 (TSST-1) that exacerbate the inflammatory response by nonspecifically activating T cells [190, 193–196]. S. aureus superantigens also can skew the cutaneous immune response towards the Th2 cytokine profile, thereby increasing S. aureus superinfection in atopic dermatitis [197]. Recently, S. aureus δ-toxin has been shown to induce allergic skin inflammation via mast cell activation in mice, providing yet another mechanism by which S. aureus contributes to the pathogenesis of atopic dermatitis [198]. Taken together, T cells likely play an important role in colonization and superinfection of atopic dermatitis lesions by increasing levels of Th2 cytokines, activation of T cells by superantigens and other toxin-mediated effects on promoting skin inflammation.
Third, recent evidence suggests that Th17 cells and IL-17 responses in humans may also be protective against S. aureus skin infections. The differentiation of naïve T cells into Th17 cells has been shown to be mediated by IL-6, IL-21, IL-23, TGFβ and/or IL-1β, which cause expression of the transcription factor RORγt in mature Th17 cells, which produce IL-17A, IL-17F, IL-21 and IL-22 [199, 200]. The strongest evidence for a protective role of Th17 cells against S. aureus skin infections comes from the study of the rare orphan disease hyper-IgE syndrome (HIES) (also called Job’s syndrome after the biblical character Job whose faithfulness was tested by an affliction with sores and boils over his entire body [201]). HIES patients suffer from an eczema-like skin eruption and chronic and recurrent skin infections with S. aureus and C. albicans [201]. HIES patients were found to have loss-of-function mutations in the signaling molecule STAT3 [202, 203], which rendered them deficient in Th17 cells [204–206]. Additionally, patients with IL-17RA or IL-17F deficiency were found to also be susceptible to C. albicans and to a lesser extent S. aureus SSTIs [207]. These findings suggest that STAT3 and IL-17/Th17 responses, in particular, play a key role in host defense against S. aureus infections in skin. However, although S. aureus-specific Th17 cells are found in blood from healthy humans [208], it remains unknown whether Th17 cells in patients without rare genetic conditions promote protection against S. aureus skin infections [209, 210].
Similarly, although initial investigations in mice suggested that Th1 responses, including IFNγ responses, promoted neutrophil recruitment against a S. aureus skin surgical wound infection [211, 212] and intravenous S. aureus infections [213–215], more recent studies have revealed that IL-17 responses in mice, like in humans, are critical for cutaneous host defense against S. aureus skin infections. In a mouse model of S. aureus skin infection, IL-17A/F produced by γδ T cells (rather than Th17 cells) was essential in producing neutrophil-attracting chemokines and granulopoiesis factors to promote neutrophil recruitment and bacterial clearance (Fig. 16.5) [216]. Interestingly, IL-17A/F in the infected skin was not induced in IL-1R- or MyD88-deficient mice, indicating that the γδ T cell IL-17A/F required IL-1R/MyD88 activation [216]. Another study found that IL-17A/F-deficient mice suffer from spontaneous S. aureus skin infections [217]. In addition, several Th17 cell-inducing vaccines (the candidal adhesion protein rAls3p-N, S. aureus clumping factor A [ClfA], S. aureus iron regulated surface determinant B [IsdB] and a UV-irradiated S. aureus vaccine) protected mice against an intravenous and/or cutaneous S. aureus challenge [213, 218, 219], suggesting that Th17 cell-inducing vaccines could potentially be effective against S. aureus cutaneous infections in humans. Interestingly, IL-20 cytokines (IL-19, IL-20 and IL-24) have been shown to enhance a S. aureus skin infection in mice by inhibiting IL-1β and IL-17 responses and treatment with an antibody against the shared receptor IL-20RB resulted in improved outcomes [220]. Finally, two recent studies have found that Th17 cells and IL-17-producing γδ T cells were protective against a recurrent S. aureus skin infection model and a S. aureus surgical wound model in mice, respectively [221, 222]. The mechanism for how anti-S. aureus Th17 are generated is an active area of investigation but a recent study found that Langerhans cells in mouse epidermis produced elevated amounts of IL-6, IL-1β, and IL-23, which promoted Th17 differentiation in response to cutaneous challenge with S. aureus and C. albicans [223]. Similarly, IL-6, IL-1β, and IL-23 also contributed to Th17 cell differentiation of S. aureus-specific Th17 cells isolated from human blood [208].
B Cell Immune Response
The B cell-mediated immune response against S. aureus involves the production of antibodies directed against specific antigenic components of S. aureus. These antibodies play an important role in opsonizing S. aureus and facilitating antibody-mediated phagocytosis by neutrophils and macrophages [209, 210]. After an acute S. aureus infection (including skin infection), antibody levels have been shown to rise, including specific antibodies against toxins (e.g., α-toxin, PVL), virulence factors (e.g., superantigens), cell-wall proteins (e.g., ClfA) and non-protein antigens (capsular polysaccharides, LTA and PGN) [224]. One study demonstrated that the antibody repertoires differed in patients with superficial versus deep-seated S. aureus skin infections [225]. Studies using various animal models of S. aureus infection have provided further evidence that antibodies against different S. aureus components can provide some level of protection against S. aureus infection [209, 210].
The importance of B cell responses and antibodies in host defense against S. aureus infections is further provided by the existence of protein A, an important virulence factor that S. aureus uses to counteract antibody-mediated responses [226]. Protein A of S. aureus binds antibody in the incorrect orientation, thus enabling S. aureus to evade antibody detection and subsequent antibody-mediated phagocytosis [226].
There have been attempts to develop vaccines and passive immunization strategies to promote antibody-mediated responses against S. aureus in humans [209, 210]. These vaccines were designed to target molecules on bacterial surfaces to enhance opsonophagocytosis such as capsular polysaccharides, ClfA and IsdB [227–229]. However, to date, all of these vaccines have failed in clinical trials [227–229]. It is unknown whether an antibody-based vaccine alone will promote durable immunity against S. aureus skin infections. However, more recent vaccines were designed to target toxins and virulence factors rather than mechanisms such as opsonophagocytosis and this may be a more efficacious approach [209, 210]. Recently, a study in children with and without recurrent S. aureus skin infections found that high natural antibodies against α-toxin correlated with protection against recurrence [230], providing rationale for these newer antibody-based vaccination strategies that target S. aureus toxins.
Group A Streptococcus
Group A Streptococcus (Streptococcus pyogenes) is a Gram-positive extracellular bacterial pathogen that causes superficial and invasive skin infections such as impetigo, erysipelas, cellulitis, scarlet fever, and necrotizing fasciitis and is the most common cause of bacterial pharyngitis (especially in children) [231, 232]. Group A Streptococcus infections can cause other severe infections such as streptococcal toxic shock syndrome, septic arthritis, osteomyelitis, septicemia, pneumonia and meningitis [231]. In addition, after a group A Streptococcus infection, immunologic-mediated diseases such as guttate psoriasis, acute rheumatic fever, and glomerulonephritis may ensue [231]. The World Health Organization estimates that there are approximately 600 million cases of noninvasive pharyngitis and 110 million skin infections caused by group A Streptococcus globally per year [231]. In the U.S., there is an estimated 8950–11,500 cases of invasive group A Streptococcus infections annually (including erysipelas, cellulitis, and necrotizing fasciitis), which result in 1050–1850 deaths [233]. Thus, group A Streptococcus continues to be a major cause of superficial and invasive skin infections both globally and in the U.S.
Clinical Manifestations
Group A Streptococcus causes superficial skin infections such as impetigo and invasive skin infections such as erysipelas, an infection of the superficial layers of the skin and cutaneous lymphatics (Fig. 16.6), or cellulitis, an infection involving the deep dermis and subcutaneous tissue [234]. Group A Streptococcus also causes necrotizing fasciitis, which is a severe skin and soft-tissue infection that results in total destruction of the deep fat and fascia and often precedes streptococcal sepsis, shock and multi-organ failure [234]. In addition, scarlet fever, which is usually associated with a streptococcal throat infection, is characterized by a morbilliform rash, strawberry tongue and desquamation of skin [231, 232]. This constellation of clinical findings in scarlet fever is caused by streptococcal pyrogenic exotoxins (Spe), especially SpeA, B and C, which act as superantigens [231, 232].
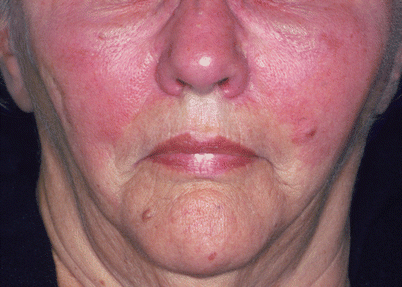
Fig. 16.6
Group A Streptococcus erysipelas of the face. The involved skin shows a sharply demarcated, erythematous, and edematous plaque (Photograph is courtesy of the Victor D. Newcomer collection at UCLA and Logical Images, Inc.)
Innate Immune Responses Against Group A Streptococcus
The innate immune response against group A Streptococcus is similar to that against S. aureus and includes soluble factors such as antimicrobial peptides and complement components, PRRs such as TLRs and NOD2 and innate immune system cells such as neutrophils and monocytes/macrophages. However, there are some key differences in the immune response against group A Streptococcus, especially with regard to the recognition and activity of M protein expressed by group A Streptococcus.
Antimicrobial Peptides
Both α- and β-defensins (HBD1-3) have direct antimicrobial activity against group A Streptococcus [235–238]. In addition, stimulation of keratinocytes with group A Streptococcus increases production of HBD2 [239]. Cathelicidin also has direct antimicrobial activity against group A Streptococcus infections in mouse models of skin infection and in cultures of human keratinocytes or mast cells [240–244]. Cathelicidin production is upregulated in wounded human or mouse skin, which protects the healing wound from infection by group A Streptococcus [245]. Thus, both defensins and cathelicidin have antimicrobial activity and play a key role in the innate immune response against group A Streptococcus.
The importance of antimicrobial peptides in the innate immune response against group A Streptococcus is further illustrated by the existence of several mechanisms that group A Streptococcus utilizes to inhibit their function. For example, group A Streptococcus produces streptococcal inhibitor of complement (SIC), which inhibits human α-defensins, HBDs 1-3, LL-37, and lysozyme [236, 238] as well as DRS (distantly related to SIC), which inhibits hBD3 [238]. SpeB can cleave and inactivate LL-37 [246]. GAS can also bind plasmin on the bacterial cell surface to facilitate degradation of LL-37 [247]. In addition, extracellular proteases released by group A Streptococcus can generate dermatan sulphate from host proteoglycans, which subsequently binds to and inactivates α-defensins [248]. Finally, similar to S. aureus, group A Streptococcus also secretes products from the dltABCD operon to reduce the negative charge of the bacterial envelope to resist the activity of LL-37 and other antimicrobial peptides [249].
Complement Activation
The importance of complement in the immune response against group A Streptococcus is illustrated by the existence of multiple factors produced by group A Streptococcus that inhibit complement activity [231]. The M protein of group A Streptococcus inhibits complement activity by several different mechanisms [250, 251]. M protein directly binds to and enhances the function of factor H (FH) and FH-like protein, host proteins that inhibit complement activation and prevent C3b-mediated phagocytosis [252–258]. In addition, a fibronectin-binding protein (Fba) was shown to have similar FH/FH-like protein binding activity as M protein to inhibit complement and enhance virulence [259–261]. Group A Streptococcus M protein also binds to and enhances the function of C4b-binding protein (C4BP), a host protein that downregulates complement activation by accelerating the decay and prevent formation of C3- and C5-convertases [262–267]. Furthermore, M protein binds fibrinogen, which inhibits complement-mediated phagocytosis by reducing the amount of C3 convertase on the surface of group A Streptococcus [268]. In addition to M protein, group A Streptococcus also secretes C5a peptidase (ScpA), which cleaves C5a and inhibits neutrophil recruitment [269–273]. Finally, the group A Streptococcus-derived protein, SIC, not only inhibits antimicrobial peptides (see above), but also binds to C5b-C7 complexes and prevents formation of the MAC [274, 275].
Pattern Recognition Receptors that Recognize Components of Group A Streptococcus
The recognition of group A Streptococcus appears to be quite different from S. aureus, despite both of them being Gram-positive bacteria. Prior work has found that macrophages and dendritic cells produced TNFα and IL-6 in a mechanism independent of TLR2, TLR4 and TLR9 [276–278]. Rather, a major host recognition mechanism for group A Streptococcus involved the induction of type I interferon (e.g., IFN-α and IFN-β), which involved DNA from group A Streptococcus to activate signaling molecules IRF3, STING, TBK1 and partially MyD88 in macrophages and streptococcal RNA triggering of IRF5 and MyD88 in dendritic cells (DCs) [278]. Furthermore, the type I interferon response was protective against a lethal subcutaneous group A Streptococcus infection model in mice [278]. Other reports found a role of TLR9 in inducing hypoxia-inducible factor-1α (HIF-1α), nitric oxide and oxidative burst in host defense and clearance of group A Streptococcus in cutaneous and systemic infection models in mice [279]. Interestingly, certain strains of Group A Streptococcus possess a bacteriophage that encodes Sda1, which is a DNase that inhibits recognition of the bacterial DNA by TLR9, thus suppressing TLR9-mediated type I interferon-mediated host defense mechanisms [280]. Other PRRs might also be involved in recognition of Group A Streptococcus. For example, cell wall fragments from group A Streptococcus induced less joint inflammation in mice deficient in NOD2 than wildtype mice and the cell wall fragments were capable of activating NOD2 expressed by human macrophages, suggesting that NOD2 is a PRR for group A Streptococcus [281]. Finally, group A Streptococcus resulted in activation of the NLRP3/ASC inflammasome to trigger caspase-1 activation and IL-1β secretion in a mechanism that was dependent upon NF-kB and the virulence factor streptolysin O but independent of exogenous ATP, P2X7R and TLR signaling [282]. Taken together, the PRRs involved in recognition of group A Streptococcus are not entirely clear (and are different than those of S. aureus), but type I interferon responses, NOD2 and NLRP3/ASC inflammasome activation have been identified to be important for host defense. It should be mentioned that the M protein of group A Streptococcus has been shown to interact with TLR2 on human monocytes leading to production of IL-6, IL-1β and TNF-α [283]. However, the M protein also binds to CD46 (membrane cofactor protein) on the surface of human keratinocytes and this interaction facilitates the ability of group A Streptococcus to invade these cells [284–287]. Thus, the M protein of group A Streptococcus might induce inflammatory responses via TLR2 while also promoting invasion of host cells and disease pathogenesis [284–287].
Cellular Innate Immune Responses Against Group A Streptococcus
Neutrophils
Neutrophil infiltrates are the characteristic of acute Group A Streptococcus infections, which is consistent with its scientific name, Streptococcus pyogenes from the Latin for ‘pus-generating’ [288]. The importance of neutrophils in host defense against group A Streptococcus is further demonstrated by the existence of numerous mechanisms that group A Streptococcus utilizes to counteract neutrophil recruitment and function [231, 288, 289]. Regarding neutrophil recruitment, group A Streptococcus not only produces the C5a peptidase (see above), but also produces another peptidase called ScpC (also called SpyCEP) that degrades CXC chemokines (including IL-8 in humans and KC and MIP2 in mice) [290]. These chemokines are critical for neutrophil recruitment to sites of infection [290].
Group A Streptococcus has developed mechanisms to inhibit both complement- and antibody-mediated phagocytosis. As mentioned above, group A Streptococcus prevents complement-mediated phagocytosis via activity of M protein and Fba. In addition, group A Streptococcus secretes endoglycosidase (EndoS), and streptococcal pyrogenic exotoxin B (SpeB) [291–295]. These bacterial factors inhibit antibody-mediated phagocytosis by hydrolyzing N-linked oligosaccharides on opsonized IgG molecules and by cleaving opsonized IgG molecules into Fab and Fc fragments, respectively [291–295]. SpeB has been shown to degrade most immunoglobulin subtypes (IgG, IgA, IgM, IgD and IgE) [293]. Recently, another endoglycosidase, EndoS2, has similar activity as EndoS to inhibit IgG-mediated phagocytosis [296]. In a mouse skin infection model, group A Streptococcus mutant strains expressing protease-inactive SpeB caused significantly less necrosis and demonstrated less efficient systemic dissemination from the initial focus of skin inoculation [297]. There are also other IgG-degrading enzymes (Ide) produced by group A Streptococcus, including IdeS (also known as Mac-1), which cleaves the lower Fc region of surface bound IgG [298] and is also a bacterial homolog of the α-subunit of the β2-integrin Mac-1 that binds to CD16 (FcγRIIIB) on phagocytes to inhibit Fc-mediated phagocytosis [299]. Mac-2 is a similar IgG-degrading enzyme, has weaker endopeptidase activity than IdeS but can competitively block FcγRII and FcγRIII to inhibit antibody-mediated phagocytosis [300]. In addition, the hyaluronic acid capsule of group A Streptococcus can act as a physical barrier to nonspecifically resist phagocytosis [301]. Group A Streptococcus also resists antibody-mediated phagocytosis by forming large bacterial aggregates via binding fibronectin and recruiting collagen fibers [302]. Taken together, group A Streptococcus produces several different factors that can inhibit both complement- and antibody-mediated phagocytosis.
There are several mechanisms that group A Streptococcus utilizes to inhibit neutrophil function. First, group A Streptococcus can directly induce neutrophil lysis or apoptosis, effectively eliminating their antimicrobial activity [303, 304]. Second, in addition to SIC, which inhibits antimicrobial peptides (see above), group A Streptococcus produces several enzymes that inhibit ROS-mediated microbicidal toxicity such as glutathione peroxidase, superoxide dismutase, alkylhydroperoxidase and alkylhydroperoxidase reductase [305–307]. Furthermore, the bacteriophage-encoded Sda1, which is produced by certain strains of Group A Streptococcus, (see above) [308, 309], as well as the nuclease SpnA [310], degrade DNA in neutrophil NETs, thus inhibiting the antimicrobial and killing mechanisms of NETs.
Adaptive Immune Responses Against Group A Streptococcus
Both B and T cells play a role in adaptive immune responses against group A Streptococcus infections. In particular, antibodies and T cells that recognize antigenic components of M protein have been shown to produce protective immune responses that prevent colonization and infection by group A Streptococcus [311–319]. Similar to S. aureus, group A Streptococcus also produces several superantigens, including streptococcal pyrogenic exotoxins (SPEs) (serotypes A, C and G to M) and the streptococcal mitogenic exotoxin SMEZn [320]. These superantigens nonspecifically activate T cells and contribute to the pathogenesis of group A Streptococcus infections [320].
The important role of adaptive immunity in host defense against group A Streptococcus has led to several different vaccination strategies to produce protective antibody responses [231, 321]. A safe human vaccine against group A Streptococcus has been challenging given the genetic diversity among clinical isolates as well as producing a vaccine that does not increase the risk for development of autoimmune sequellae such as acute rheumatic fever and acute poststreptococcal glomerulonephritis [231, 321, 322]. Since antibodies against M protein of group A Streptococcus have been shown to offer protection against colonization and infection, several vaccines have targeted different antigenic epitopes of the M protein, including 26- and 30-valent N terminal domain vaccines [314, 315] as well as vaccines targeting more conserved epitopes [316–319]. In addition, other vaccine approaches have been attempted, including vaccines directed against other streptococcal antigens, including the group A carbohydrate, C5a peptidase, fibronectin-binding protein A (FbaA), fibronectin-binding protein 54 (Fbp54), streptococcal protective antigen (Spa), SpeB, SpeC, serine protease (SpyCEP), serine esterase (SSe) and several other antigens [231, 321, 322]. These strategies have had varying successes in animal studies; whether the efficacy of these vaccines will translate to humans is not yet known. As newer technologies are becoming more available, such as high-throughput proteomics approaches [323–326], defining candidate vaccine targets with enhanced efficacy and broad strain coverage will greatly help in the development of a successful human vaccine against group A Streptococcus infections in the future.
Conclusion
Recent discoveries involving innate and adaptive immune responses against S. aureus and group A Streptococcus have greatly improved our understanding of these common skin infections. As antimicrobial resistance is creating a serious threat to public health, novel strategies to enhance protective host immune mechanisms against bacterial skin infections represent an alternative approach to combat these infections while minimizing antibiotic resistance. The mechanisms of cutaneous host defense and bacterial pathogenesis will be important factors to consider for the development of future immunotherapies and vaccine strategies against these common bacterial skin pathogens.
Questions
- 1.
Get Clinical Tree app for offline access
Staphylococcus aureus is a common cause of all of the following skin infections, EXCEPT:
- A.
CellulitisRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



- A.