Fig. 22.1
Spectrum of atopic/eczematous dermatitis. List of genes in which mutations can cause eczematous dermatitis associated with staphylococcal colonization bacterial and fungal infections, and other clinical manifestations such as food allergy and asthma
Table 22.1
AD-like genetic disorders
Disease | Causative gene | Staphylococcal infections |
|---|---|---|
Netherton syndrome | SPINK5 | Skin |
Peeling skin disease | CDSN | Skin |
SAM syndrome | DSG1 | N.D. |
Autosomal dominant hyper-IgE syndrome | STAT3 | Skin and respiratory tract |
Autosomal recessive hyper-IgE syndrome | DOCK8 | Skin and respiratory tract |
Wiskott-Aldrich syndrome | WAS | Skin and respiratory tract |
ADAM17 deficiency | ADAM17 | Skin |
IPEX syndrome | FOXP3 | Skin and respiratory tract |
Netherton syndrome is an autosomal recessive skin disorder characterized by congenital ichthyosis and severe atopic manifestations including eczematous lesions and staphylococcal skin colonization, food allergy and rhinitis with high serum IgE. It is caused by mutations in SPINK5, encoding the serine protease inhibitor LEKTI [21]. LEKTI inhibits several members of kallikreins (KLK) including KLK5, KLK7 and KLK14, the balance of which is crucial to regulate desquamation. Loss of LEKTI leads to aberrant enhancement of multiple proteolytic events in epidermis, and Stimulates proinflammatory signals via protease activated receptor 2 (PAR2). Spink5-deficient mice and KLK5-overexpressed mice manifest Netherton syndrome-like, as well as AD-like phenotypes, such as eczematous inflammation, and high serum IgE and TSLP [22–24], which was attributed to PAR2-mediated TSLP up-regulation [23]. A significant association of a SPINK5 polymorphism with AD has been found, indicating an overlap of disease pathogenesis between NS and AD [25].
A generalized, inflammatory type of peeling skin syndrome (type B) also manifests as eczematous dermatitis and food allergies. This disease is associated with mutations in CDSN, which leads to a complete loss of corneodesmosin [26]. Corneodesmosin is an adhesion glycoprotein located in corneodesmosomes, and deletion of Cdsn resulted in hair and skin abnormalities also in mice [27].
Recently, loss-of-function mutations of another important transmembrane structural protein have been reported in patients with eczematous dermatitis. Homozygous mutations in DSG1, which encodes desmoglein1, a major constituent of desmosomes, cause a syndrome featuring severe dermatitis, multiple allergies and metabolic wasting (SAM syndrome) [28].
Interestingly, like FLG, gene defects in the above diseases and those listed in Table 22.1 directly impair normal differentiation and formation of the upper epidermis to the stratum corneum. Furthermore, it is interesting to note that disease affecting lower parts of the epidermis, such as epidermolysis bullosa, does not result in eczematous dermatitis or asthma and food allergies despite the formation of ulcers or erosions that potentially allow antigen penetration. Existence of upper epidermis ~ stratum corneum with dysfunctional barrier appears crucial for the development of AD and subsequent atopic march.
AD-Like Genetic Disorders with Immunodeficiency
Patients with primary immunodeficiencies, which are caused by mutations in genes mainly associated with the development and regulation of immune cells, can also exhibit eczematous dermatitis that closely resembles AD. An autosomal dominant form of hyper-IgE syndrome (HIES) is caused by mutations in STAT3, and manifests as eczematous dermatitis with recurrent staphylococcal infections [29, 30]. Staphlycococcal infections not only occur in skin and soft tissue, but also in the respiratory tract. Patients with an autosomal recessive type of HIES caused by DOCK8 mutations also have eczema and recurrent staphylococcal skin infections [31]. Another rare primary immunodeficiency, Wiscot-Aldrich syndrome, is characterized by eczematous dermatitis with recurrent skin infections, in addition to thrombocytopenia [32]. It is caused by mutations in WAS, a protein that is important for proper actin cytoskeleton function during cell motility, and is expressed in most hematopoietic cells.
The hallmark of the above-mentioned diseases is immune dysregulation that leads to increased susceptibility for recurrent infections in skin as well as other organs. Gene mutations responsible for these diseases have not been implicated to be involved in epidermal differentiation. Although it is clear that barrier dysfunction can lead to AD, a pathway that arises from immune dysfunction to barrier dysfunction is also possible. AD-like genetic diseases that result from barrier dysfunction and immunodeficiency might represent opposite ends of the AD spectrum.
A Gene Mutation That Affects Both Immunity and Barrier Formation
A new genetic disorder has been reported that could be fitted within the atopic spectrum. Patients with loss-of-function mutations in ADAM17 manifest eczematous dermatitis and pustules with Staphylococcus aureus skin infections [33]. The function of ADAM17 in skin has been investigated in murine studies, which demonstrated that deficiency of ADAM17 down-stream signaling pathways such as EGFR or Notch are responsible for impaired barrier integrity, immune dysregulation and staphylococcal dysbiosis [34–36]. Note that many of these diseases in the atopic disease spectrum have staphyloccoccal skin colonization in common [37], which may be responsible for the induction of eczematous dermatitis.
Pathomechanisms
Pathophysiology of AD is complex and, as suggested in the above section, multiple pathways can lead to one common clinical phenotype. There are three major drivers that contribute to the pathogenesis of AD: (1) Impaired epidermal barrier (2) immunological abnormalities and (3) altered skin microbiota. Immunological disturbance as the primary cause of AD has been widely investigated because the mainstream of AD has been considered as Th2 type allergic responses against foreign antigens. High serum IgE concentration and eosinophilia are main laboratory features that support the Th2 concept. Barrier function in AD has been a point of focus over the past several years, which gained huge momentum after the discovery of FLG mutations. Meanwhile, increased colonization of S. aureus in skin of patients with AD has long been known, but was difficult to explain in the context of allergic inflammation. Recent advances in microbiome research are beginning to provide better understanding of interactions between the microbiome and host immunity and highlight the importance of S. aureus colonization in AD pathogenesis (Fig. 22.2).
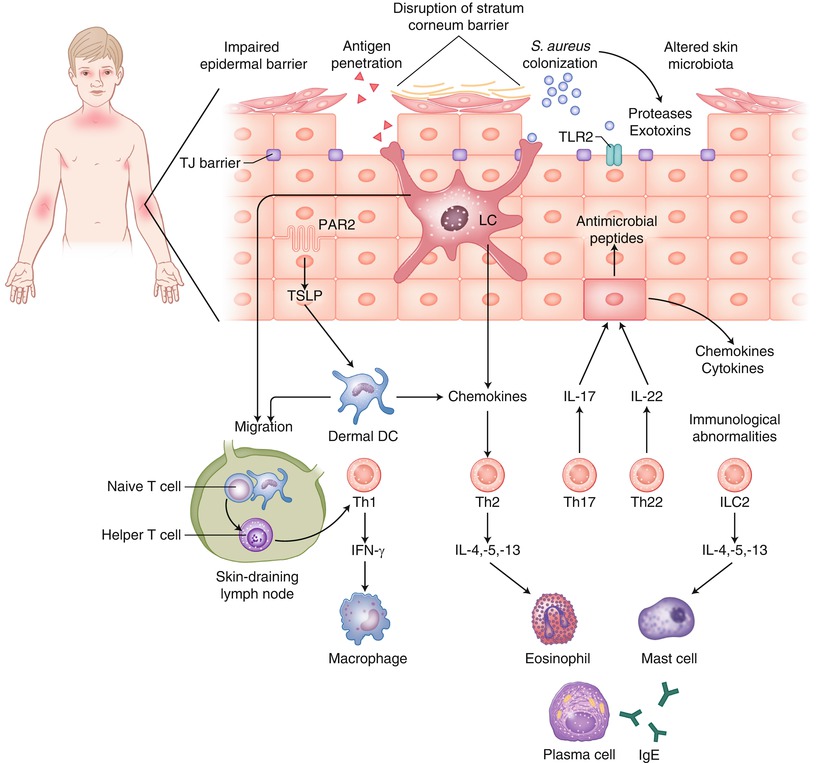
Fig. 22.2
Three major driving forces for the pathogenesis of atopic dermatitis: Impaired epidermal barrier, immunological abnormalities and altered skin microbiota. Loss of barrier integrity is believed to lead to antigen penetration and initiates inflammatory responses driven by a mixed type of cytokine producing T cells. Although the contribution of altered skin microbiota is not yet conclusive, staphylococcus aureus colonization is universal in AD as well as other genetic dermatosis and immunodeficiencies that exhibit eczematous dermatitis
Impaired Epidermal Barrier
Dry skin is caused by barrier alteration followed by increased trans epidermal water loss, and is considered an important underlying condition in AD. Since loss-of-function mutations in FLG have been reported in patients with AD, the manner in which filaggrin defects contribute to barrier dysfunction has been intensively studied. Given the fact that filaggrin is a major structural protein in the stratum corneum, filaggrin insufficiency would be expected to result in a number of structural, biophysical and functional changes, and in aggregate result in impaired barrier formation. Filaggrin breakdown products form natural moisturizing factors (NMF), which are thought to be essential for stratum corneum hydration. Although it is widely accepted that FLG mutations result in outside-in barrier dysfunction, the mechanism of this defect is yet to be fully elucidated.
Better understanding of filaggrin biology has been gained from murine models of filaggrin deficiency. The spontaneous mutant flaky tail/matted mice exhibit eczematous inflammation and enhanced immune response to cutaneous allergen exposure. A 1-bp deletion mutation of Flg was found [38], enforcing the previous finding in humans. Further studies investigating flaky tail (Flg ft ) mice showed clinical AD-like manifestations with barrier abnormalities and Th17-dominated skin inflammation [39–41]. Those studies suggested a crucial role of filaggrin in the maintenance of physical epidermal barrier. Unexpectedly, however, genomic deletion of filaggrin in mice led to altered stratum corneum barrier and enhanced percutaneous sensitization, but did not lead to the onset of spontaneous dermatitis under specific pathogen free conditions [42], demonstrating that filaggrin deficiency alone is not sufficient to induce eczematous dermatitis. In fact, recent studies revealed that the matted (ma) mutation, but not Flg mutation, is responsible for the dermatitis phenotype in Flg ft mice [43, 44], which raises the question of whether eczematous inflammation is truly a result of enhanced antigen penetration subsequent to barrier dysfunction.
Nevertheless, filaggrin is also reported to play an important role in maintaining pH balance of SC. Acidic conditions caused by filaggrin breakdown products, urocanic acid and pyrrolidone carboxylic acid, have been reported to inhibit the growth of S.aureus [45]. Moreover, reduced SC NMF components and consequent increased SC pH result in enhanced cleavage of proinflammatory IL-1 cytokines, which were increased in AD patients with FLG mutations as well as filaggrin deficient Flg ft/ft mice [46]. In light of the matted mutant mice and Flg deficient mice, it remains to be clarified whether Flg mutation alone is responsible for the above findings.
Immunological Abnormalities
Th2 and Th1
Based on the clinical observation that patients with AD often manifest blood eosinophilia and elevated serum IgE levels, AD was long considered to be a Th2 type allergic disease. Th2 cells produce IL-4, IL-5 and IL-13 and activate eosinophils, basophils and mast cells as well as IgE-producing B cells, which are all involved in allergic reactions. In the 1990s, two studies detected mRNA expression of cytokines via in situ hybridization and reported increased expression of Th2 cytokines in skin of AD patients [47, 48]. In addition, the serum level of the Th2-attracting chemokine, CCL17/TARC, correlates with AD activity [49], indicating contribution of Th2 type response in the disease flare. In contrast, elevation of Th1 cytokines, such as IFN-γ and IL-2, has also been detected in the late phase in patch test reactions of AD patients [50]. Those studies supported the evidence of Th2 cytokine bias as well as involvement of Th1 cytokines in AD.
Findings on TSLP have provided new insights into the mechanism of Th2 biased-pathogenesis in AD. TSLP is a potent activator of dendritic cells and plays an important role in the induction of CD4+ T cell-mediated allergic inflammation. TSLP promotes DCs to induce Th2-chemokines and consequently enhances the production of Th2 cytokines from T cells. TSLP was highly expressed in epidermis of AD patients [51]. Murine models of epidermis-specific overexpression of TSLP (K5-TSLP transgenic mouse) developed T cell-mediated dermatitis [52]. Other murine studies showed that TSLP promotes basophil responses and Th2 cytokine-mediated inflammation [53], and that TSLP induction through PAR2 activation induces dermatitis and basophil accumulation in flaky tail mice [54].
Although a number of studies have been conducted to prove Th2 involvement in the pathogenesis of AD, therapeutic targeting of Th2-mediating factors has not proven effective. Neutralization of Th2 cytokines or IgE and inactivation of B cells has not yielded promising outcomes. True clinical relevance of Th2 has yet to be demonstrated.
Interestingly, epithelial cells directly regulate cutaneous sensory neurons via TSLP activation to promote pruritus, which is dependent on PAR2-triggering Ca influx and NFAT translocation [55], suggesting the contribution of a non-immunological mechanism mediated by TSLP in AD.
Th17 and Th22
Emerging reports suggest the contribution of Th17 and Th22 in atopic skin [56]. IL-17 and IL-22 function to accelerate innate immune responses against extracellular pathogens and are linked to many immune/autoimmune related diseases. IL-17 and IL-22 are overexpressed in skin of AD [57, 58], and infiltrations of Th17 and Th22 cells into lesional skin have been reported [59, 60]. Correlation of serum IL-22 with CCL17 [61] and increase of IL-13/IL-22 producing cells in AD [62] suggest a link between Th22 and Th2 cytokines.
Th17-associated skin inflammation can be observed in several mouse models with AD-like inflammation, such as the flaky tail mice [40], transgenic kallikrein 5 mice [24] and Adam17 conditional knockout mice [36]. Importantly, IL-17 and IL-22 synergistically promote the induction of antimicrobial peptides from keratinocytes [63], and S. aureus-derived enterotoxin induces IL-17 production of T cells isolated from AD patients [64], suggesting that production of IL-17 and IL-22 is associated with colonization of S. aureus in AD skin (Table 22.2).
Table 22.2
Mouse models of AD
Mouse strain | Characteristics | References |
|---|---|---|
Spontaneously developing models | ||
Nc/Nga | Scratch, increased TEWL, dermatitis, high IgE and increased expression of Th2 cytokines in skin | |
Flaky tail (ma/ma, Flg ft/ft ) | Scratch, increased TEWL, dermatitis, high IgE and enhanced percutaneous sensitization to OVA | |
Genetically engineered models | ||
IL-4 transgenic | Scratch, dermatitis, S. aureus colonization and high IgE | [128] |
IL-13 transgenic | Scratch, dermatitis, high IgE and increased expression of Th2 cytokines in skin | [129] |
IL-31 transgenic | Scratch and dermatitis | [67] |
TSLP transgenic | Dermatitis, high IgE and increased production of Th2 cytokines | [52] |
CASP1 transgenic | Scratch, dermatitis and high IgE | [65] |
Foxp3 knockout | Dermatitis and high IgE | [130] |
ADAM17 conditional knockout | Scratch, high IgE, increased TEWL, S. aureus colonization and increased production of Th2 and Th17 cytokines | |
EGFR conditional knockout | Scratch, high IgE, increased TEWL, S. aureus colonization and increased production of Th2 and Th17 cytokines | |
Notch1 and 2 conditional knockout | Dermatitis, high IgE and increased expression of Th2 cytokines in skin | [131] |
Epicutaneous sensitization models | ||
OVA sensitized | Scratch, dermatitis, high IgE, increased expression of Th2 cytokines and chemokines in skin | |
Hapten induced | Th2-associated dermatitis and high IgE | [134] |
Other Cytokines
Overexpression of IL-18, an IL-1 superfamily, in murine epidermis resulted in dermatitis with elevated serum IgE and Th2 cytokines [65]. Epidermal IL-18 levels of patients with AD was correlated with SCORAD and prevalence of S. aureus colonization [66]. Transgenic mice overexpressing IL-31, an IL-6 family cytokine that is produced by Th2 cells, developed pruritic dermatitis [67]. Serum IL-31 is also increased in atopic individuals and the production of IL-31 in human PBMCs can be induced by staphylococcal enterotoxin B [68].
Innate Lymphoid Cells
After their recent discovery, innate lymphoid cells (ILCs) have emerged as essential effectors of innate immunity and potent players in the pathogenesis of many inflammatory diseases. ILCs resemble lymphocytes in morphology and function as effector cells through the production of several cytokines, but lack a T cell receptors. ILCs are divided into three groups based on their producing cytokines. Group 2 ILCs produce type 2 cytokines (IL-4, IL-5, IL-9, IL-13) and are implicated in allergic immune responses. The first report in the context of AD revealed that ILC2 are enriched in human AD skin lesions and in mouse skin with Vitamin D3-induced inflammation, and that ILC2 responses were dependent on TSLP [69]. Another report also found accumulation of ILC2 in lesional skin of atopic patients as well as in skin of house dust mite-exposed murine skin with dependency on IL-25 and IL-33 [70]. IL-25 and IL-33 are inducers of ILC2 expansion, and in fact, transgenic mice with epidermis-specific IL-33 overexpression developed AD-like skin lesions with increase of ILC2 [71]. Consistently, the expression of IL-33 receptor is increased in human AD skin [72].
Regulatory T Cells
Regulatory T cells (Treg) are CD25+, Foxp3+ T cell subsets, which play important roles in suppressing T cell responses. Although it is generally accepted that Treg modulate allergic inflammation by suppressing the Th2 response, the exact role of Treg in AD pathogenesis still remains unclear [73]. Treg was absent in AD lesional skin in one study [74], while it existed in other studies [75, 76]. Mutations of the Treg master regulator gene, FOXP3, result in the absent or dysfunctional Tregs and cause IPEX syndrome. Importantly, some of IPEX syndrome patients manifest allergic disorders including asthma, food allergy and eczema [77, 78], suggesting a strong link between the loss of Tregs and the onset of allergic diseases.
Dendritic Cells
Specialized antigen-presenting cells (APCs), or dendritic cells (DCs), play pivotal roles in the initiation of immune reactions through activation and modulation of T cells. Although the contribution of Langerhans’ cells (LC) in initiating humoral responses against protein antigens that were taken up through the epidermal tight junctions have been established in mice [79–81], the role(s) of DCs in AD pathogenesis still remains enigmatic. It has been reported that human epidermal LCs express the high affinity receptor for IgE (FceRI) [82]. In addition, inflammatory DCs, that expressed CD206, CD209 and FceRI, but did not contain Birbeck granules, were found in lesional epidermis of AD patients, and were termed ‘inflammatory dendritic epidermal cells’ (IDEC) [83]. IDECs can be found to infiltrate not only epidermis, but also dermis of AD lesions [84].
FceRI on DCs mediate IgE-facilitated allergen presentation [85]. It is therefore possible that FceRI expressed on surfaces of LCs and IDECs are involved in IgE-mediated allergic reactions also in AD skin. TSLP receptor deficient mice reconstituted with normal bone marrow (therefore keratinocytes and LCs are the only relevant cells that lack TSLP receptors) resulted in decrease of clinical manifestations and OVA-specific IgE upon epicutaneous OVA sensitization in mice [86]. Moreover, vitamin D3 analogue-induced eczematous inflammation was decreased in the absence of LCs in mice [87]. Those experimental settings provide evidence that LCs contribute to Th2 as well as eczematous responses in mice, and further suggest that they may contribute to AD pathogenesis.
Altered Skin Microbiota
Skin harbors a diverse community of microorganisms, referred to as the microbiota. Composition of microbiota is greatly influenced by the condition of skin, and possibly, vice versa. Skin resident microbes are implicated in cutaneous immune system development and function and pathology of skin diseases. It has been long known that skin of patients with AD is heavily colonized with S. aureus [88]. Density of S. aureus colonization correlated with SCORAD [89, 90], suggesting the contribution of S. aureus to the mechanism of the disease flare.
Recent technological advances on next generation DNA sequencing have allowed the utilization of culture independent metagenomic approaches to investigate the collective genomes of resident bacteria, the microbiome, and have uncovered the details of bacterial community structure in skin [91]. Bacterial 16S rRNA sequencing analysis in children with AD revealed temporal shifts in skin microbiome during flares, in which bacterial diversity decreases, and S. aureus predominates [92]. A microbiome study in patients with primary immunodeficiencies featuring AD-like eczema revealed that microbial compositions were altered in patients compared with controls, and prevalence of Staphylococcus and Corynebacteirum were positively correlated with disease severity [37]. Consistent with previously conducted studies with culture dependent detection techniques, these recent studies confirmed the emergence of S. aureus in eczematous lesions and provided new insight into bidirectional influences between microbes, particularly S. aureus, and atopic skin. However, the cause-and-effect relationship between staphylococcal colonization and eczema formation is still inconclusive, and the clinical studies provide evidence for anti-bacterial therapies only for secondary infections of eczematous lesions, but not for the treatment of eczema itself.
S. aureus is commonly found in healthy human skin, but could cause staphylococcal skin infections such as impetigo, folliculitis and abscesses under certain conditions. S. aureus produces a wide array of virulence factors, and it is not clear yet whether such factors contribute to eczema formation. Nevertheless, it is easily imaginable that virulence factors could have direct or indirect contributions, via host immune responses, to eczema formation or aggravation. A major group of virulence factors of S. aureus, such as proteases, hyaluronidase, lipase and nuclease, comprise extracellular enzymes. Another group is a family of exotoxins including cytolytic membrane-damaging toxins (mainly α, β, γ, and δ-toxins and Panton-Valentine leukocidin) and superantigens such as staphylococcal enterotoxin (SEA and SEB), toxic shock syndrome toxin-1 and exfoliative toxin A and B. An important characteristic of S. aureus is its ability to secrete exotoxins that disrupt host cell membrane. The correlation between the colonization of exotoxin-producing S. aureus strains and disease severity of AD has been reported [93].
The mechanism by which superantigens contribute to disease flares in AD has not been completely elucidated. Several reports have suggested effects of staphylococcal exotoxins on immune cells, by activating T cells by specific interaction between TCR and the toxin [94, 95], and by promoting allergic reactions through IgE generated against staphylococcal exotoxins [96–98]. A recent report showed that mast cell degranulation is induced by δ-toxin, suggesting pathological role(s) of secreted toxins in allergic skin disease [99].
Why S. aureus is capable of abundantly colonizing skin of AD patients is still another matter of debate, and might represent important mechanisms that could affect future therapeutic strategies. Skin is equipped with innate mechanisms that prevent overgrowth of S. aureus. Such mechanisms include protection against adherence of S. aureus to corneocytes. Acidic conditioning of skin inhibits the growth of S. aureus, and the production of antimicrobial peptide (AMP) by keratinocytes, sebocytes and eccrine glands likely targets S. aureus. Neutrophil responses are also promoted by IL-1β produced by keratinocytes [100]. It is possible that one or several of these factors are impaired in skin of AD patients.
One of the initial immune responses against bacterial pathogens is innate immunity via the recognition of pathogen-associated molecular patterns of bacteria through pathogen related receptors, and the most studied family of such receptors are Toll-like receptors (TLRs). Genetic variants of TLR2 have been studied in the context of AD, because it recognizes cell-wall products of S. aureus, and its polymorphism has been reported in AD patients with a history of S.aureus infections and increased serum IgE [101, 102].
AMPs are evolutionarily ancient innate immune effectors and are thought to contribute in immune defense of epithelial surface. Expression levels of AMPs have been reported in several studies, but the results of those are controversial. Reduced expression of cathelicidins (LL-37) and human β-defensin 2 (HBD-2) in AD skin was first reported in comparison to psoriatic skin [103], and was attributed to Th2 cytokine milieu in AD [104]. However, follow-up studies revealed the expressions of AMPs in lesional skin of AD are not actually decreased, but rather increased, when compared with healthy control skin [105] [90]. Meanwhile, mobilization of HBD-3 appears to be inhibited in keratinocytes derived from AD patients [106].
Increasing number of studies in the recent years has shown that imbalances in the composition of the intestinal microbiota are associated with both intestinal and extra-intestinal diseases. The association of altered gut microbiota with an increased risk of developing AD has been reported in several studies [12, 107–109]. Efforts have been made to “normalize” the imbalance of microbial composition in the intestine via the supplementation of probiotics. A double-blind, randomized placebo-controlled clinical trial demonstrated that taking Lactobacillus GG has a preventative effect in children at high risk of AD [110]. In the era of microbiome, such studies should be corroborated with microbiome analysis via next generation sequencing.
Treatment
Management of AD requires multiple approaches, targeting various aspects of pathogenesis. The cores of such aspects that were discussed in this chapter are barrier disruption, inflammation and, for future perspectives, dysbiosis. While strategic development of novel therapeutics will heavily depend on basic science, treatment strategies at the patient level should be determined based on clinical conditions of individual patients as well as evidence that is provided by randomized controlled trials (RCTs), optimally supported by data from basic and clinical research.
Dry skin is a fundamental clinical element in AD, and the importance of SC barrier has been highlighted after the discovery of FLG mutations in patients with AD. Skin care aimed to maintain barrier integrity, thereby preventing epicutaneous sensitization against environmental allergens, would be a fundamental therapeutic strategy that accommodates most, if not all, disease phases in AD. There is strong evidence that supporting skin hydration by topical application of moisturizers reduces disease severity and prevents flares [111]. Pharmacologic interventions that directly target filaggrin expression that enforce barrier function may be a potential therapeutic approach in the future.
Control of inflammation by the use of anti-inflammatory or immunomodulating agents is another mainstream therapeutic strategy. Topical corticosteroids and calcineurin inhibitors are recommended based on evidence provided by a number of RCTs [111–113]. Systemic application of cyclosporin A is effective but only indicated for patients with AD refractory to conventional topical treatment [114, 115].
Although a large amount of scientific data on immunological pathophysiology of AD has been accumulated, specific therapies for inhibiting specific types of immune cells or cytokines are being studied in clinical trials. The results of clinical trials with new biologics such as monoclonal anti-IgE antibody (Omalizumab) and a chimeric monoclonal antibody against B-cell surface antigen CD20 (Rituximab) had been rather disappointing, although effective in a fraction of patients [112, 116]. Some early clinical trials have shown beneficial effects of recombinant IFN-γ in patients with severe AD [117, 118], but the level of evidence is still of limited-quality. With the variety of helper T cell subsets that have become known, it might be worth revisiting whether modulation and deviation of certain helper T cell subset(s) is a relevant strategy in the treatment of AD.
Inducing immunologic tolerance against specific allergens is an ultimate goal for the treatment of IgE-mediated allergic disorders in which major antigens have been established. There is enough evidence to support the effectiveness of allergen immunotherapy for the treatment of allergic rhinitis, allergic conjunctivitis and allergic asthma, but evidence for AD is still limited [119].
Consistent with epidemiological data that prevalence of AD negatively correlates with exposure time to UV, efficacy of phototherapy has been noted [120]. UV therapy is well established to exert immunosuppressive effects in vivo, for example by increasing IL-10 production [121], and can be utilized to attenuate skin inflammation. Meanwhile, specific recommendation of UV for a definitive AD condition has not been made, and its contraindication in patients treated with tacrolimus renders it to be considered a second line treatment.
In addition to initial culture-based studies, microbiome studies in humans have established that S. aureus dominates the microbiota during AD flares [92]. Consistently, it has been demonstrated in epidermal Adam17-deficient mice that S. aureus is capable of driving eczematous dermatitis, and that intense antibiotic cocktail targeting S. aureus nearly eradicates eczematous inflammation [36]. The concept that eczematous dermatitis is driven by S. aureus offers novel and attractive therapeutic strategies. However, despite the evidence of S. aureus colonization, and despite the longstanding interest in targeting this bacteria to try to improve AD symptoms, the efficacy of anti-bacterial treatment for AD patients remains unclear.
A Cochrane review of RCTs found a lack of quality trials to support the use of anti-bacterial therapies, for both topical and systemic antibiotics [122]. If antibiotics are to be utilized, well-designed randomized studies need to be conducted, considering the presence of Staphylococcal dysbiosis. With the current available evidence, the recommendation for the use of systemic antibiotics for AD patients is limited to those manifesting clear clinical signs of bacterial infections [120], and not for mere colonization. Intensified antibody cocktails might be necessary to target colonizing S. aureus, as have been done experimentally in mice. On the contrary, sustained systemic exposure to antibiotics will have adverse effects at distant sites such as the gut [123, 124], thus the approach utilizing systemic antibiotics might not be of practical use. In this context, the use of bleach baths is recommended for moderate to severe AD [125]. It is possible that bleach baths might be targeting Staphylococcal dysbiosis, and such skin microbiome-directed local therapy that normalizes dysbiosis long term, is awaited.
Questions
- 1.
Which of the following genes associated with AD/AD-like genetic disorders have function(s) in barrier formation?
- A.
DOCK8
- B.
FLG
- C.
CDSN
- D.
WAS
- E.
All of the above
- A.
- 2.
Get Clinical Tree app for offline access
Which of the following genes associated with AD/AD-like genetic disorders have function(s) in immunity?
- A.
STAT3
- B.
DSG1
- C.
SPINK5
- D.
FOXP3Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



- A.