Atopic dermatitis (AD) is increasingly recognized as a complex, inflammatory skin disease involving interplay of multiple elements. This article notes key advances in understanding of immune dysregulation, skin barrier dysfunction, environmental, genetic, and microbial influences orchestrating disease pathogenesis, and the relevance of therapeutic interventions in each area. Accumulating evidence and the discovery of new T-cell subsets has matured AD as a multiple-cytokine-axes–driven disorder, evolved from the widely held belief of it being a biphasic Th1/Th2 disease. These new insights have led to active trials testing multiple, targeted therapeutics with better efficacy and safety-profiles.
Key points
- •
Atopic dermatitis (AD) is increasingly recognized as multifactorial and heterogeneous with differing molecular or cellular phenotypes characterizing different populations.
- •
Accumulating studies continue clarifying key interactions among susceptibility genes, environmental factors, microbiome, impaired barrier integrity, and immune dysregulation.
- •
Identification of immune subsets, including Th17, Th22, and Th9, has shifted disease paradigms from biphasic Th1/Th2 driven to a complex, multi-axes disease.
- •
Advances in AD pathomechanisms are leading to robust development of novel, targeted therapeutics, whereas current treatments are limited and may harbor toxic effects.
Introduction
Atopic dermatitis (AD) is recognized as a multifactorial, heterogeneous disease characterized by different clinical phenotypes based on interactions of susceptibility genes, environmental factors, impaired skin barrier integrity, and immune dysregulation. Although barrier impairment and immune dysregulation play major roles in pathogenesis, their sequential order is unclear. The outside-in theory suggests that dysfunctional epidermal barrier incites the disease with secondary immunologic changes. Conversely, the inside-out hypothesis holds that immune dysregulation drives the disease and barrier changes are an epiphenomenon. Recent genome-wide association studies identified loci correlated with autoimmune regulation, including genes associated with regulation of innate host defenses and T-cell function, linking AD to other autoimmune or inflammatory diseases. Present treatments are limited and are not without adverse effects, creating a large unmet need for targeted approaches.
This article highlights key advances in the understanding of AD pathophysiology, including immune dysregulation, skin barrier dysfunction, environmental, genetic, and microbiome effects, with implications for therapeutic interventions.
Introduction
Atopic dermatitis (AD) is recognized as a multifactorial, heterogeneous disease characterized by different clinical phenotypes based on interactions of susceptibility genes, environmental factors, impaired skin barrier integrity, and immune dysregulation. Although barrier impairment and immune dysregulation play major roles in pathogenesis, their sequential order is unclear. The outside-in theory suggests that dysfunctional epidermal barrier incites the disease with secondary immunologic changes. Conversely, the inside-out hypothesis holds that immune dysregulation drives the disease and barrier changes are an epiphenomenon. Recent genome-wide association studies identified loci correlated with autoimmune regulation, including genes associated with regulation of innate host defenses and T-cell function, linking AD to other autoimmune or inflammatory diseases. Present treatments are limited and are not without adverse effects, creating a large unmet need for targeted approaches.
This article highlights key advances in the understanding of AD pathophysiology, including immune dysregulation, skin barrier dysfunction, environmental, genetic, and microbiome effects, with implications for therapeutic interventions.
Immune dysregulation
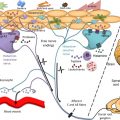
Dysregulation of both innate and adaptive immune systems are involved in AD. Although keratinocytes, antimicrobial peptides (AMPs), innate lymphoid cells group 2 (ILC-2), and toll-like receptors (TLRs) are major players of the innate arm, the discovery of T helper (Th) subsets Th17/Th22 has shed new light on the adaptive arm of AD pathogenesis, progressing from past models perceiving AD as a Th1/Th2 biphasic disease into the current concept of a multiple-cytokine axes disorder.
Although the pathogenetic role of autoimmunity in AD remains to be elucidated, a recent meta-analysis reported autoimmune phenomenon in up to 91% of AD patients, possibly a consequence of unrecognized self-epitopes.
Innate Immune System
Critical by virtue of their location, keratinocytes serve as sentinel cells with various downstream effects. AMPs, generated by keratinocytes, are divided into 2 classes: cathelicidin (LL-37) and human-β-defensins 2 and 3, which play key roles in pathogen clearance and maintenance of tight junction integrity. In AD skin, both subsets are reduced, contributing to increased infections. AMPs induce several cytokines/chemokines, including interleukin (IL)-4, IL-13, IL-25, IL-33, and thymic stromal lymphopoietin (TSLP). IL-25, IL-33, and TSLP affect dermal ILC-2 to produce IL-5 and IL-13. ILC-2 increases and subsequent Th2 cytokine production, leads to Th2 axis augmentation in positive feedback loops. Mutations in TLRs and nucleotide-binding oligomerization domain-like receptors (NLRs) also play roles in AD. TLR2, which enhances tight junction integrity and defenses against Staphylococcus aureus and herpes simplex virus infections, shows decreased expression in AD.
Adaptive Immune System
T helper-1
Expanded in chronic lesions, Th1 has been shown in severe AD patients to have a skin selective or cutaneous lymphocyte antigen (CLA)-positive defect which might potentially add to susceptibility for cutaneous infections. Ustekinumab, an IL-12/IL-23p40 antagonist, is currently being explored in clinical trials ( Table 1 ). Mixed results have been documented, with some case reports showing clinical efficacy, whereas others demonstrated only limited benefit in clearing AD lesions. A recent phase II, double-blinded, placebo-controlled study showed ustekinumab to modulate Th1, Th17, Th22, and also Th2-related AD genes; however, there were no significant differences in clinical outcomes versus placebo. The interpretation of these results is limited due to the crossover study design, added to the effect of topical corticosteroids, which were allowed in all cohorts. A more recent study of severe AD subjects found ustekinumab to reduce Eczema Area and Severity Index (EASI) by 50% at the end of treatment. In this study, ustekinumab also decreased epidermal hyperplasia or proliferation, dermal T-cell infiltrates, dendritic cells (DCs), and mast cells with quantitative polymerase chain reaction showing reduction in Th2/Th22 markers. These conflicting results demand larger trials.
| Category or Targeted Axis | Target | Trade Name and ClinicalTrials.gov Identifier |
|---|---|---|
| Th1/Th17 | Anti-IL-12/23 (anti-p40) mAb | Stelara (ustekinumab)/ NCT01806662 |
| Anti-IL-17 mAb | Secukinumab/ NCT02594098 | |
| Th2 | Anti-IL-4Rα mAb | Dupilumab/ NCT02260986 |
| Anti-IL-13 mAb | NCT02340234 NCT02347176 | |
| CRTH2 antagonist | NCT01785602 NCT02002208 NCT02590289 | |
| Th22 | Anti-IL-22 mAb | ILV-094/ NCT01941537 |
| TSLP antagonist | Tezepelumab/ NCT02525094 | |
| PDE4 inhibitors | Otezla (Apremilast)/ NCT02087943 NCT01856764 NCT01461941 NCT02094235 NCT02068352 NCT01993420 NCT01037881 | |
| Crisaborole/ NCT02118792 NCT02118766 | ||
| Antipruritic | Anti-IL-31 mAb | NCT01986933 NCT01614756 |
| Antitropomyosin receptor kinase A | NCT01808157 | |
| Antineurokinin | NCT02004041 NCT01033097 | |
| Prostaglandin D2 receptor agonist | NCT00914186 | |
| Anti-TRPV1 channel | NCT02583022 | |
| Kappa-opioid receptor agonist | NCT02475447 NCT02576093 |
T helper-2
Increased Th2-related marker expression, including IL-5, IL-13, IL-10, IL-31, and chemokine (C-C motif) ligand (CCL)-5, CCL13, and CCL18, is prominent in acute lesions and augmented in chronic AD. Th2 products downregulate AMP and epidermal differentiation complex (EDC) genes, thus suppressing major terminal differentiation proteins (filaggrin [FLG], loricrin [LOR], and involucrin [IVL]), whereas upregulating kallikreins (KLKs) are responsible for corneodesmosome degradation. Th2 products permit antibody responses that include immunoglobulin (Ig)-E isotype-switching and mast cell or eosinophil differentiation, though IgE itself is not a key mediator of AD pathogenesis. IL-33/IL-31, involved in pruritus induction and in food allergies, were found to be upregulated in AD lesional skin. These new insights have led to active testing of immune axes-specific drugs. Dupilumab, an anti-IL-4Rα, has been shown to normalize Th2 inflammatory cytokines but also reverse barrier abnormalities, underlining the ongoing crosstalk between these 2 components (see Table 1 ).
T helper-9
Th9 cells are a relatively newly recognized skin-tropic T-cell subset that are generated from naïve T cells (in the presence of transforming growth factor [TGF]-β and IL-4) or differentiated from Th2 cells. Although Th9 cells are thought to be main producers of IL-9, which is elevated in AD skin lesions and sera of adults and children, the role of Th9/IL-9 in AD pathophysiology is obscure. IL-9 functions to drive T-cell survival, proliferation, and secretion of inflammatory mediators, and seems to play a role in activation of ILCs, in which it enhances IL-5 and IL-13 production. In keratinocytes, IL-9 induces VEGF, which has been associated with epidermal changes seen in AD. Nonetheless, a genomic and molecular profiling of nonlesional, acute, and chronic AD lesions showed no significant difference in IL-9 levels between these tissues, though acute and chronic lesions had similar increases in IL-9 levels compared with nonlesional skin. Other studies show IL-9 levels in skin and in serum to correlate with Scoring Atopic Dermatitis (SCORAD), serum IgE, and CCL17 levels. However, the relevance of Th9/IL-9 in AD pathogenesis remains incompletely understood and is only beginning elucidation.
T helper-22
Th22 and associated markers are upregulated in acute AD with intensification in chronic lesions. IL-22 was reported to correlate with disease severity, induce epidermal hyperplasia, inhibit terminal differentiation, and regulate skin barrier function. Drugs targeting the Th22 axis are actively undergoing phase II trials (see Table 1 ).
T helper-17
The Th17 immune axis is generally attenuated in AD, though some studies report it being increased. Although the role of Th17 T cells in AD remains controversial, it is thought that the robust Th2 cytokine signal seen in AD inhibits the Th17 axis. IL-17 has been shown to infiltrate AD lesions, modify epithelial cells and keratinocytes to produce inflammatory cytokines, and downregulate filaggrin and other epidermal barrier gene expressions. Anti-IL17 phase II trials are ongoing (see Table 1 ).
Dendritic Cells
CD11 + myeloid DCs (mDCs), are found in chronic lesions. Other DC subtypes include plasmacytoid DCs, increased in chronic AD, and blood DC antigen positive and CD123 + DCs, which secrete interferon (IFN)-α and mediate lesion formation. Inflammatory dendritic epidermal DCs (IDECs), a type of mDC, are important contributors to T-cell activation. Dermal DCs (dDCs), which express the same markers as IDECs, are localized to the dermis and have important immunostimulatory effects in acute AD. dDCs express high levels of TSLP, which induces a Th1/Th17 response and Th2 polarization through various chemoattractants (CCL17, CCL 22, CCL24) in an OX40-dependent mechanism.
Regulatory T cells
Reports show increased regulatory T cell (Treg) counts in AD lesions, and serum Treg levels to positively correlate with disease severity. However, the exact role of Tregs in AD remains to be clarified.
Finally, increased activity of phosphodiesterase (PDE) activity was implicated in AD, reducing Th1 and enhancing Th2 signals. Inhibition of PDE4, located in keratinocytes and a wide variety of immune cells, increases levels of cAMP, leading to inhibition of proinflammatory cytokines. PDE4 inhibition by apremilast and crisaborole, have shown to be efficacious (see Table 1 ).
Certain patient groups show distinct immune responses, supporting the complex molecular and cellular phenotype characterizing AD. Asian AD is Th17 polarized, whereas intrinsic AD has more robust immune activation and a significantly increased Th17 signal. Pediatric AD is characterized by a multi-axes activation in skin lesions but only Th2/Th1 imbalance in blood. These findings implicate tailored strategies that target distinct phenotypes.
Skin barrier
The epidermal barrier prevents allergen penetration, maintains skin hydration, and displays antimicrobial activity, while having an ongoing interaction with the adaptive immune system. The barrier is composed of a complex matrix of structural proteins and lipids held together by desmosomes and tight junctions. Deficiency of these components, lack of keratinocyte differentiation, and immune dysregulation are the basis for barrier dysfunction in AD.
Filaggrin
The skin barrier is composed of a cornified envelope (CE), essential for barrier function. The CE is composed of terminally differentiated keratinocytes, structural proteins (LOR, IVL, and FLG), and lipids. FLG is a structural protein important for cornification, skin hydration, and AMP function. FLG mutations are strongly associated with AD but are found only in 15% to 50% of patients. Additionally, approximately 40% of FLG mutations carriers never develop AD. FLG deficiency is associated with early onset, severe AD, greater allergen sensitization, and increased susceptibility to infections. FLG degradation products are components of the natural moisturizing factor (NMF), vital for skin hydration. Lower levels of NMF and greater transepidermal water loss (TEWL) have been observed in AD patients with FLG mutations.
Lipids
Lesional and nonlesional AD skin is characterized by reduced ceramide composition, as well as reduced ceramide to cholesterol ratios, alterations which lead to increased TEWL. Cole and colleagues performed a meta-analysis to define the AD transcriptome showing abnormalities in lipid metabolism genes despite normal FLG genes. Additionally, Th2 activation correlated with downregulation of key epidermal lipids. Ceramide-dominant moisturizers have shown to restore integrity of the skin barrier. A recent evidence-based review evaluating the efficacy of topical or oral oils concluded that topical sunflower seed or coconut oil may be potential therapies for AD ; however, larger, well-designed studies are needed.
Tight Junctions
Tight junctions, transmembrane proteins regulating the permeability of the epidermis, are decreased in AD. Claudins are tight junction proteins found on keratinocytes that further strengthen the epidermal barrier. Claudin1 gene expression is downregulated in AD, and shown to negatively correlate with Th2 cytokines, serum IgE, and serum eosinophils. Yuki and colleagues reported that impaired tight junctions affect lipids and FLG processing by disturbing skin pH. TLR2 activation, reduced in AD, has been shown to increase expression of tight junction proteins and enhance barrier function.
Skin pH
An acidic skin pH (4–6) is necessary to maintain the integrity of the stratum corneum, lipid metabolism, epidermal differentiation, and antimicrobial functions. The baseline skin pH in AD is high. FLG deficiency may contribute to this increased pH due to decreased acidic FLG breakdown products. Increased pH enhances serine protease activity in the stratum corneum. Jang and colleagues evaluated the role of pH in AD pathogenesis and found that in mice, skin alkalinization-induced KLK5 led to skin barrier dysfunction, pruritus, and dermatitis. The increased activity of serine proteases leads to over-degradation of structural proteins and lipids, and decreased barrier integrity. Acidic pH inhibits the growth of pathogens such as S aureus , and decreases the expression of staphylococcal surface-binding proteins. Thus, alkaline pH in AD promotes bacterial growth, contributing to an altered skin microbial profile.
Skin barrier plays a major role in AD pathogenesis, making it an attractive prophylactic and therapeutic target. Early use of emollients can prevent AD in 50% of high-risk infants, highlighting the critical role barrier integrity plays in AD initiation.
The interplay between barrier integrity and immune dysregulation is complex ( Fig. 1 ). For example, Th2 and Th22 downregulate EDC gene expression, suppress terminal differentiation, inhibit desmoglein 3 expression and ceramide synthesis, modulate FLG expression, and induce epidermal hyperplasia. Additionally, correction of the barrier ameliorates inflammation, reinforcing this concept.