Dermatofibrosarcoma protuberans (DFSP) is a rare soft-tissue tumor that most commonly presents on the trunk and extremities of adults. It is characterized by low metastatic potential and a favorable prognosis, but extensive subclinical growth can contribute to a high risk of local recurrence. Surgical excision is the first-line treatment, using Mohs micrographic surgery or wide local excision with careful evaluation of the peripheral and deep surgical margins. Adjuvant therapy may be beneficial in patients with unresectable, recurrent, or metastatic DFSP. Historically, adjuvant radiation therapy has been used to reduce the risk of local recurrence when residual disease is present after surgery. The advent of targeted molecular therapies, such as the selective tyrosine kinase inhibitor, imatinib mesylate, has provided new effective and safe options for adjuvant treatment of DFSP.
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, low-grade soft tissue neoplasm accounting for less than approximately 0.1% of all cancers and 1% of all soft tissue sarcomas. The overall annual incidence of DFSP is 4.2 per million of all cancers as reported in the Surveillance, Epidemiology, and End Results (SEER) cancer registries. The incidence rate is higher among blacks compared with other groups (6.5 cases per million population), but the incidence among whites has been slowly increasing over the past 30 years, in part due to improved diagnostic immunohistochemical techniques. DFSP develops at approximately equal rates between women and men except in older individuals (>70 years), in whom men have a higher incidence.
Clinical presentation
DFSP growth is characteristically indolent. These tumors enlarge gradually over a period of years, but may present with extensive subclinical invasion into underlying subcutaneous tissue, fascia, muscle, or even bone. The broadly infiltrative nature of DFSP contributes to its high rate of local recurrence following treatment with standard surgical excision.
Although DFSP can occur congenitally or in childhood, it most commonly presents in adults between the ages of 30 and 50. Fewer than 10% of DFSPs are diagnosed before the age of 20, and the incidence of this tumor in childhood may in fact be underestimated, since the diagnosis is often delayed. Congenital DFSP can have a variable presentation and may be challenging to definitively remove if the diagnosis is not made early or if scarring from incomplete excision is present.
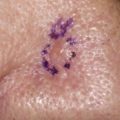
DFSP classically appears as a violaceous or erythematous nodular plaque ( Fig. 1 ) but can also feature skin-colored, erythematous, brown-tinged, or yellow-tinged areas within patches, nodules, or plaques. Areas of induration, telangiectasia, or atrophy may be evident at presentation or may appear over time. Its clinical appearance can vary from indistinct small plaques to large, exophytic tumors that can bleed or ulcerate. Early lesions tend to be asymptomatic but can become painful over months to years due to deeper tissue invasion or accelerated growth. Development of DFSP or acceleration of its growth has been associated with trauma and scars, including sites of vaccination, as well as pregnancy. While predominantly located on the trunk (42%) and extremities (34%), DFSP may also occur on the head and neck, and in these locations it is associated with a greater risk of morbidity and local recurrence.
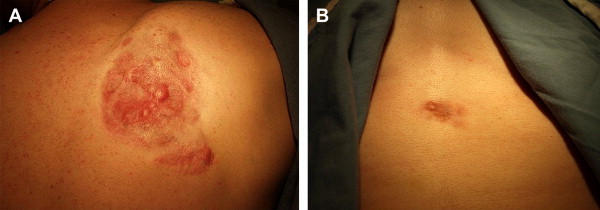
Together with its often-benign clinical appearance, the rare nature of DFSP and its tendency for indolent growth can prompt diagnostic challenges. Clinically, DFSP can be mistaken for hypertrophic or keloidal scarring, morphea, epidermoid cysts, melanoma, or metastatic neoplasms. In congenital DFSP, early lesions may be difficult to distinguish from vascular malformations, infantile fibromatosis or myofibromatosis, fibrosarcoma, or fibrous hamartoma.
Staging and prognosis
A definitive staging system that can assist with prediction of patient outcomes does not yet exist for DFSP. In some cases, the Short German Guidelines or the general American Musculoskeletal Tumor Society Staging System, which take into account high- or low-grade histopathology, local tumor extension, or distant spread, may be helpful, but the 5-year survival for classical DFSP is over 99%. Imaging studies for staging purposes are typically not required, since tumor involvement is most frequently limited to local disease. If metastasis occurs, it spreads most frequently to regional lymph nodes. While distant metastasis occurs in less than 5% of cases, it is associated with a poor prognosis, with death from widespread metastatic disease typically occurring within 2 years. In advanced, recurrent, or high-grade variants of DFSP, the risk of hematogenous dissemination is greater and most commonly leads to pulmonary metastases. Imaging of the chest using computed tomography (CT) to evaluate for pulmonary metastasis is therefore indicated in high-risk clinical situations. Other potential sites of hematogenous spread include bone, brain, heart, and other soft tissues.
Distant metastasis and disease-specific mortality are usually consequences of local recurrence after inadequate surgical excision. In some prospective case series, disease-free survival after wide local excision correlated inversely with tumor depth, tumor grade, patient age, positive margin after primary resection, or presence of the high-risk fibrosarcomatous variant on histology.
Staging and prognosis
A definitive staging system that can assist with prediction of patient outcomes does not yet exist for DFSP. In some cases, the Short German Guidelines or the general American Musculoskeletal Tumor Society Staging System, which take into account high- or low-grade histopathology, local tumor extension, or distant spread, may be helpful, but the 5-year survival for classical DFSP is over 99%. Imaging studies for staging purposes are typically not required, since tumor involvement is most frequently limited to local disease. If metastasis occurs, it spreads most frequently to regional lymph nodes. While distant metastasis occurs in less than 5% of cases, it is associated with a poor prognosis, with death from widespread metastatic disease typically occurring within 2 years. In advanced, recurrent, or high-grade variants of DFSP, the risk of hematogenous dissemination is greater and most commonly leads to pulmonary metastases. Imaging of the chest using computed tomography (CT) to evaluate for pulmonary metastasis is therefore indicated in high-risk clinical situations. Other potential sites of hematogenous spread include bone, brain, heart, and other soft tissues.
Distant metastasis and disease-specific mortality are usually consequences of local recurrence after inadequate surgical excision. In some prospective case series, disease-free survival after wide local excision correlated inversely with tumor depth, tumor grade, patient age, positive margin after primary resection, or presence of the high-risk fibrosarcomatous variant on histology.
Histopathology and immunohistochemistry
Clinical suspicion for DFSP following a complete skin examination typically requires an incisional skin biopsy to help confirm the diagnosis. Histological features suggestive of DFSP typically include a dense collection of monomorphous fusiform cells forming focal storiform or cartwheel configurations as demonstrated in Fig. 2 A . Early lesions may feature an area of dermal sparing, or a Grenz zone, which is clearly seen just beneath the epidermis. Coursing through the dermis and infiltrating the subcutaneous fat (see Fig. 2 B), an extensive proliferation of spindle cells disrupts the adipose tissue architecture and creates a honeycomb or lace-like appearance. Deeper projections into fascia or muscle can further complicate demarcation of the tumor border and subsequent surgical management. Determination of the precise surgical margin in DFSP can be challenging owing to the tumor’s bland histologic appearance and diffuse infiltration, as well as its similarity to other neoplasms. Occasionally DFSP can be difficult to distinguish from dermatofibroma, dermatomyofibroma, fibrosarcoma, leiomyoscarcoma, malignant fibrous histiocytoma (also called pleomorphic scarcoma), or atypical fibroxanthoma. Dermatofibromas have a tendency to be better demarcated and to have less infiltrative subdermal peripheral extension compared with DFSP. Malignant fibrous histiocytoma and atypical fibroxanthoma display greater mitotic activity and pleomorphism than DFSP.
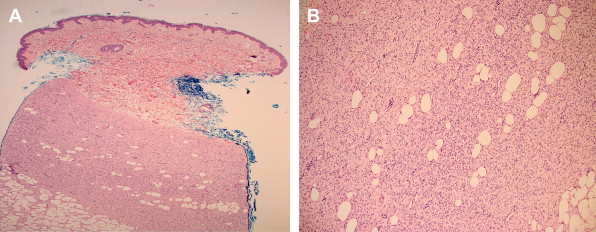
Approximately 10% of DFSPs transform into a high-grade fibrosarcomatous variant with increased cellularity, mitosis, and pleomorphic spindle cells in a herringbone configuration deep in the dermis. Other uncommon variants of DFSP include pigmented DFSP, also called Bednar tumor, which occurs in approximately 1% of patients, possibly with a higher incidence among blacks. DFSPs with myofibroblastic, myxoid, and neurofibromatous differentiation have been rarely reported. The giant cell fibroblastoma variant demonstrates areas with spindle-shaped cells in a myxoid background with characteristic multinucleated giant cells.
Additional immunohistochemical markers may be used to improve diagnostic accuracy when required. Besides human hematopoietic progenitor cells, CD34 antigen is expressed by a subpopulation of dermal dendritic cells and has been shown to be selectively expressed in DFSP. Nodular or fibrosarcomatous areas of DFSP may exhibit variable expression of CD34, reducing the sensitivity of this marker. CD34 can help to differentiate DFSP from dermatofibroma. In many cases, DFSP stains positively for CD34 and negatively for factor XIIIa, whereas dermatofibroma is CD34-negative and factor XIIIa-positive. Neural markers (S-100) can help distinguish DFSP from desmoplastic melanoma, neurofibroma, schwannoma, and malignant peripheral nerve shealth tumor. Rarely, DFSP with neurofibromatous changes may be focally positive for immunostaining with S-100. The cytoplasmic histiocyte immunohistochemical marker, CD68, selectively stains for malignant fibrous histiocytoma and atypical fibroxanthoma, but not for DFSP, facilitating the distinction of these other two spindle cell tumors from DFSP.
Pathogenesis of DFSP
DFSP is thought to originate from cutaneous mesenchymal cells. In accordance with the cancer stem cell hypothesis, mutations in multipotent cutaneous mesenchymal stem cells residing in the connective tissue sheath or hair follicle papillae putatively induce formation of DFSP neoplasms. Advances in understanding the molecular pathogenesis of DFSP have enabled positive identification of most of these tumors using modern molecular diagnostic techniques. Characteristic cytogenetic chromosomal alterations can now be detected in over 90% of patients with DFSP. Aberrations in chromosomes 17 and 22 were first identified in DFSP in 1990 ( Fig. 3 A ). At the cytogenetic level, DFSP harbors either supernumerary ring chromosomes or translocations that are often unbalanced. The supernumerary ring chromosomes combine portions of chromosome 17 with chromosome 22 in a circular formation (see Fig. 3 B). Instead of forming rings, t(17;22) translocations connect chromosomes 17 and 22 in a linear manner (see Fig. 3 C). Karyotype analysis using fluorescence in situ hybridization (FISH) can detect these chromosomal anomalies within DFSP cells in tissue samples. In case series, supernumerary ring chromosomes are more common in adult DFSP cases while translocations predominate in the pediatric population.
Juxtaposition of chromosomes 17 and 22 in both rings and translocations results in oncogenic fusion of the type 1 alpha I collagen gene (COL1A1) and the beta chain of platelet-derived growth factor (PDGFB) gene. Under normal circumstances, active transcription of the COL1A1 gene produces type 1 collagen, the most abundant protein in the body. Aberrant linkage of the genetic elements that usually drive expression of the COL1A1 gene with that of the PDGFB gene results in amplified expression of PDGFB protein. Overproduction of PDGFB leads to autocrine or paracrine stimulation of its cellular receptor, the PDGF receptor (PDGFR), a cell–surface receptor tyrosine kinase. Since PDGFB serves as a potent cellular growth factor, its overexpression is thought to contribute to tumorigenesis. The COL1A1-PDGFB fusion has also been shown to be present in several uncommon variants including pigmented DFSP (Bednar tumor). The chimeric COL1A1-PDGFB gene fusion can be detected at the DNA level using polymerase chain reaction (PCR) technology, which serves as another diagnostic tool for analyzing tissue samples suspicious for DFSP. Although useful in diagnostic testing, the cytogenetic aberrations in chromosomes 17 and 22 that characterize most DFSP tumors have not been shown to influence or predict clinical outcomes. There is no correlation between COL1A1 breakpoints on chromosome 17 and any particular clinical or histopathologic characteristics.
Management of DFSP: overview
Since DFSP tumors exhibit unpredictable and widespread subclinical extension, surgical excision with comprehensive margin evaluation prior to reconstruction is the optimal treatment of choice for DFSP. When a surgical margin is positive or narrow without the possibility of further surgery, use of adjuvant therapies might salvage local control. Alternatives or adjuncts to surgical management include radiation therapy and imatinib (Gleevec) for advanced, recurrent, or metastatic DFSP. Conventional chemotherapy is considered generally ineffective. The uncommon incidence of DFSP precludes development of therapeutic strategies based on clinical evidence from randomized data or comparative studies assessing treatment modalities in parallel. Consensus treatment guidelines developed by the National Comprehensive Cancer Network derive from published case series featuring predominantly retrospective analyses. A basic algorithm for surgical management of DFSP is diagrammed in Fig. 4 . Because DFSP recurrence after years and decades has been a concern, at least semiannual clinical examination for observation is recommended, especially in the first 3 years following surgery, during which 80% of recurrences are thought to occur.
Management of DFSP: surgical excision
The deeply infiltrative nature of DFSP necessitates excision at least to the level of the underlying fascia. In addition to physical examination for fixation to deeper structures and the presence or absence of regional lymphadenopathy, presurgical imaging using magnetic resonance imaging (MRI) or CT if bony involvement is suspected can help define the extent of tumor spread.
The lowest DFSP recurrence rates have been demonstrated following excision using the Mohs micrographic surgery (MMS) technique or wide local excision (WLE) combined with complete circumferential peripheral and deep margin assessment (CPPDMA). Immunostaining with CD34 is sometimes used adjunctively during specimen processing. In the MMS procedure, the excised specimen is uniquely processed for microscopic examination using fresh tissue that is frozen and sliced horizontally or tangentially into serial sections for complete margin evaluation ( Fig. 5 A ). The entire peripheral epidermal edge and tissue depth can be visualized in the horizontal plane, since MMS excision allows for relative flattening of the surgical specimen. The process is repeated, often the same day, until the surgical margin is clear. Alternatively, a permanently fixed specimen excised using wide excision can be processed for CPPDMA or full-margin analysis. As depicted in Fig. 5 B, the perimeter of the specimen undergoes en face tangential sectioning while the residual central portion is sectioned horizontally at the base. Closure of the surgical wound may be delayed if re-excision is needed to remove any residual tumor. These thorough methods for tissue processing yield superior margin assessment, with 5 year recurrence rates as low as 0–1.7%. When wide excision may not be feasible, as in locations on the head and neck or distal extremities (featured in Fig. 6 A and B, respectively), MMS offers greater tissue-sparing potential in addition to the meticulous margin control, which is critical for minimizing likelihood of recurrence. Wide excision with traditional vertical sectioning or bread loafing (see Fig. 5 C) produces transverse slices through the excised specimen with the epidermal edge positioned above its adjacent subdermal structures. This approach represents a random sampling in which a minimum of 5% of the surgical margin is examined, leaving a bulk of the margin unchecked, with risk of false-negative interpretation and tumor regrowth.
Related posts:
 Mohs Micrographic Surgery Technique
Mohs Micrographic Surgery Technique
 Mohs Surgery for Squamous Cell Carcinoma
Mohs Surgery for Squamous Cell Carcinoma
 Management of Unusual Cutaneous Malignancies: Atypical Fibroxanthoma, Malignant Fibrous Histiocytoma, Sebaceous Carcinoma, Extramammary Paget Disease
Management of Unusual Cutaneous Malignancies: Atypical Fibroxanthoma, Malignant Fibrous Histiocytoma, Sebaceous Carcinoma, Extramammary Paget Disease
 Management of Skin Cancer in Solid-organ Transplant Recipients: A Multidisciplinary Approach
Special Considerations for Mohs Micrographic Surgery on the Eyelids, Lips, Genitalia, and Nail Unit
Multidisciplinary Approach to Large Cutaneous Tumors of the Head and Neck
Management of Skin Cancer in Solid-organ Transplant Recipients: A Multidisciplinary Approach
Special Considerations for Mohs Micrographic Surgery on the Eyelids, Lips, Genitalia, and Nail Unit
Multidisciplinary Approach to Large Cutaneous Tumors of the Head and Neck
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


