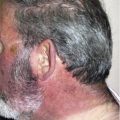
Actinic prurigo is a chronic photodermatosis with onset in childhood or before 20 years of age. It is most prevalent in Amerindians and Latin American mestizos, although it has been reported worldwide. Patients present with photodistributed, erythematous excoriated papules, cheilitis, and conjunctivitis. There is strong association with human leukocyte antigen DR4, especially the DRB1*0407 subtype. Treatment consists of photoprotection and the use of thalidomide.
Key points
- •
Actinic prurigo is a chronic photosensitivity disorder, which is more prevalent in Amerindians and Latin American mestizos and has a strong association with human leukocyte antigen DR4, especially the DRB1*0407 subtype.
- •
Clinical features are typical, but sometimes could be similar to a persistent form of polymorphic light eruption or photosensitive atopic dermatitis.
- •
Biopsies of lips and conjunctivae, in conjunction with clinical findings, help to confirm the diagnosis.
- •
Repetitive exposure to ultraviolet A and B could reproduce lesions.
- •
Thalidomide is the treatment of choice but should be used with caution, especially in women of childbearing age.
Introduction
Actinic prurigo (AP) is an uncommon, immunologically mediated photosensitivity disorder, usually beginning in childhood or before 20 years of age, although it can start later in life. It is characterized by a chronic course, with development of intensely pruritic papules, plaques, and nodules, mainly over sun-exposed skin. Involvement of lips and conjunctivae is frequent in North American and Latin American patients, and sometimes can be the only feature of the disease. Human leukocyte antigen (HLA) DR4, especially the HLA DRB1*0407 subtype, is associated with most cases of AP, suggesting an autoimmune basis for this disorder.
Some investigators, based on cases seen in the United Kingdom, consider AP as a clinical variant of polymorphic light eruption with a different genetic background. However, because of its unique clinical presentation, most Latin American dermatologists consider AP to be a distinct entity.
Because of the intense pruritus that can lead to scarring, and owing to the need for sun avoidance, AP has a great impact on the quality of life of affected patients (Dermatology Life Quality Index scores >10 [moderate effect]).
History
Robert Willan (1798) was the first author to describe a skin disease caused by sun, which he called “eczema solaris.” This report was probably the earliest of patients suffering from AP. However, most investigators consider Jonathan Hutchinson’s report about “summer prurigo” in 1878 as the primary reference to this disease, although the cases he described did not correspond completely with all clinical features of AP.
In 1956, Robert Brandt was the first to identify familial involvement in AP patients after staying in a Navajo reservation and finding individuals of this community with clinical features of AP, which he called “solar prurigo.”
The first use of the term “actinic prurigo” was by Londoño from Colombia, who used it in a 1960 publication in Spanish. He reported a series of 31 cases, establishing the frequent eczema-like component of AP, which should be differentiated from photoallergic contact dermatitis mediated by photosensitizers. In 1966, Londoño and colleagues also pointed out the possible familial character of this disease in 6 families.
In 1977 Calnan and Meara were the first English-speaking investigators to use the current term actinic prurigo to refer to a group of British patients who had the disease; they stated that AP should be clearly differentiated from polymorphic light eruption.
Even before Londoño, other Spanish-speaking investigators made important contributions to the characterization of AP: López González, an Argentinian dermatologist, presented 3 cases of this disease in 1950 under the name prurigo de verano ; Escalona, in the first edition of his Dermatology textbook (1954), described the clinical features of prurigo solar . Other investigators from Central and South America studied the disease and used different names to identify AP: syndrome cutáneo guatemalense (Cordero, 1960), dermatitis solar (Saúl, 1972), dermatitis polimorfa a la luz (Corrales Padilla, 1973), erupción polimorfa lumínica, tipo prúrigo (Hojyo and Dominguez, 1975), and prurigo solar de altiplanicie (Flores, 1975).
Introduction
Actinic prurigo (AP) is an uncommon, immunologically mediated photosensitivity disorder, usually beginning in childhood or before 20 years of age, although it can start later in life. It is characterized by a chronic course, with development of intensely pruritic papules, plaques, and nodules, mainly over sun-exposed skin. Involvement of lips and conjunctivae is frequent in North American and Latin American patients, and sometimes can be the only feature of the disease. Human leukocyte antigen (HLA) DR4, especially the HLA DRB1*0407 subtype, is associated with most cases of AP, suggesting an autoimmune basis for this disorder.
Some investigators, based on cases seen in the United Kingdom, consider AP as a clinical variant of polymorphic light eruption with a different genetic background. However, because of its unique clinical presentation, most Latin American dermatologists consider AP to be a distinct entity.
Because of the intense pruritus that can lead to scarring, and owing to the need for sun avoidance, AP has a great impact on the quality of life of affected patients (Dermatology Life Quality Index scores >10 [moderate effect]).
History
Robert Willan (1798) was the first author to describe a skin disease caused by sun, which he called “eczema solaris.” This report was probably the earliest of patients suffering from AP. However, most investigators consider Jonathan Hutchinson’s report about “summer prurigo” in 1878 as the primary reference to this disease, although the cases he described did not correspond completely with all clinical features of AP.
In 1956, Robert Brandt was the first to identify familial involvement in AP patients after staying in a Navajo reservation and finding individuals of this community with clinical features of AP, which he called “solar prurigo.”
The first use of the term “actinic prurigo” was by Londoño from Colombia, who used it in a 1960 publication in Spanish. He reported a series of 31 cases, establishing the frequent eczema-like component of AP, which should be differentiated from photoallergic contact dermatitis mediated by photosensitizers. In 1966, Londoño and colleagues also pointed out the possible familial character of this disease in 6 families.
In 1977 Calnan and Meara were the first English-speaking investigators to use the current term actinic prurigo to refer to a group of British patients who had the disease; they stated that AP should be clearly differentiated from polymorphic light eruption.
Even before Londoño, other Spanish-speaking investigators made important contributions to the characterization of AP: López González, an Argentinian dermatologist, presented 3 cases of this disease in 1950 under the name prurigo de verano ; Escalona, in the first edition of his Dermatology textbook (1954), described the clinical features of prurigo solar . Other investigators from Central and South America studied the disease and used different names to identify AP: syndrome cutáneo guatemalense (Cordero, 1960), dermatitis solar (Saúl, 1972), dermatitis polimorfa a la luz (Corrales Padilla, 1973), erupción polimorfa lumínica, tipo prúrigo (Hojyo and Dominguez, 1975), and prurigo solar de altiplanicie (Flores, 1975).
Epidemiology
AP predominantly affects indigenous tribes from North, Central, and South America, and Latin American mestizos (individuals with a mixed Caucasian and Amerindian ancestry), particularly those from Mexico, Colombia, Peru, Bolivia, Ecuador, Guatemala, Honduras, and northern Argentina. It is far less frequently seen in Europe (United Kingdom, France, Albania, Greece, Germany ), Oceania (Australia ), and Asia (Thailand, Singapore, Japan ).
Prevalence of AP varies depending on the studied population; in Canadian Aboriginals it is 0.1%, in Mexican mestizos 1.3% to 3.5%, in Trujillo (Peru) 3.4%, in Chimila Indians of Colombia 8%, and in Scotland 0.003%. This disease probably represents less than 5% of referrals to photodermatology clinics.
A family history is commonly found in closed communities; it has been reported in 75% of American Indian cases, 50% in the United Kingdom, and 4.3% to 25% in Mexico. Some investigators have found personal and family history of atopy in AP patients, even though sometimes it is difficult to clearly differentiate between photoaggravated atopic dermatitis and AP. The disease predominates in skin phototypes III to V, and is more common in women than in men with a ratio of between 2:1 and 4:1 ; notable exceptions are the Chimila indigenous tribe of Colombia and in Asians, in whom more men are affected. The effects of 17β-estradiol in preventing ultraviolet radiation (UVR)-induced immunosuppression may explain why the disease is more frequent in females, as has been suggested in polymorphic light eruption, another immunologically mediated photodermatosis that is more common in women.
AP is usually described in Latin American patients living at high altitudes (>1000 m), but has also been reported in patients residing at sea level in Colombia, Canada, and Peru. Most of the patients from high-altitude locations improve when they move to lower-altitude locales.
Pathogenesis
The pathogenesis of AP is unknown, but is clearly related to sun exposure as reflected by the distribution of the skin lesions, the differences in the behavior of the disease between the summer and winter, and the reproduction of lesions with artificial UVR sources. At present, the most accepted theory on the pathogenesis of AP is a delayed hypersensitivity reaction to an unidentified autoantigen induced by UVR, occurring in genetically susceptible individuals. This hypothesis is supported by observations that there are activated CD4-positive T cells and memory T lymphocytes in the infiltrate of the biopsies of AP patients, there is abnormal reactivity of AP lymphocytes against ultraviolet (UV)-irradiated keratinocytes, there is higher autoantibody reactivity on the skin, and there are more intense proliferative responses to isolated autologous skin antigens in AP patients in comparison with controls.
Torres-Alvarez and colleagues showed persistence of Langerhans cells in the epidermis of AP patients on UV exposure, whereas in healthy individuals these cells decrease in number in the irradiated skin. This result suggests that there is persistence of antigen-presenting cells, which could result in enhanced inflammatory response and resistance to UVR-induced immunosuppression. A study comparing the density of Langerhans cells in lesional and nonlesional skin showed that there was a lower density of Langerhans cells in lesional skin. Arrese and colleagues found high tumor necrosis factor α (TNFα) immunoreactivity in keratinocytes in the suprabasal layer of AP patients. Therefore, they proposed that UVR in subjects genetically predisposed to AP could trigger excessive TNFα production by these cells, leading to the development of lesions.
Association with HLA
There is a significant association of AP with HLA subtypes. The first studies were made with HLA class I antigens ( Table 1 ), then with class II antigens. A strong association was found with HLA DR4 ( Table 2 ), particularly with the subtype DRB1*0407 ( Tables 3 and 4 ). Menagé and colleagues postulate that HLA antigens may modify the patient’s response to sunlight-induced antigens, and may contribute to the expression of the disease. It should be noted that HLA association is not essential for the development of AP lesions.
| Country | Allele | Patients (%) | Controls (%) | OR | 95% CI |
|---|---|---|---|---|---|
| Colombia | Cw4 | 53.5 | 21 | 4.3 | 1.8–10.2 |
| B40 | 41.9 | 13 | 5.2 | 2.1–13.5 | |
| Mexico | A28 | 86.2 | 23 | 20.9 | 8.31–48.5 |
| B39 | 72.4 | 28 | 6.7 | 3.11–13.2 | |
| Canada | Cw4 | 62.5 | 25 | 5.0 | 1.4–14.6 |
| A24 | 65.6 | 21.9 | 6.8 | 2.2–20.7 |
| Country | n | Patients (%) | n | Controls (%) | OR | 95% CI |
|---|---|---|---|---|---|---|
| Mexico | 29 | 92.8 | 100 | 57 | 10.1 | 2.3–91.8 |
| Colombia | 40 | 97.5 | 40 | 35 | 8.2 | 5–13.5 |
| Great Britain | 26 | 100 | 177 | 39.5 | — | — |
| Great Britain | 66 | 80 | 126 | 39 | — | — |
| Scotland | 24 | 96 | — | — | — | — |
| Australia | 21 | 85.7 | — | — | — | — |
| Allele | Patients Allele (%) | Controls Allele (%) | OR | 95% CI | |
|---|---|---|---|---|---|
| Mexico | DRB1*0407 | 60.5 | 10.6 | 12.9 | 6.4–26 |
| Colombia | DRB1*0407 | 63.8 | 14.5 | 9.9 | 4.3–23.3 |
| Canada: Inuit | DRB1*14 | 51.2 | 26.2 | 3.0 | 1.19–7.86 |
| Country | No. of Patients | % | No. of Controls | % |
|---|---|---|---|---|
| Great Britain | 20 | 60 | 20 | 0 |
| 66 | 56 | 126 | 2 | |
| Scotland | 18 | 72 | — | — |
| Australia | 21 | 71.4 | — | — |
Whereas HLA DRB1*0407 is commonly observed in the Americas, it is uncommon in European Caucasians (4.4%–6.7% of DR4-positive individuals) and in other regions of the world, as described by Solberg and colleagues. The geographic distribution of this allele may explain why AP is more common in some regions of North, Central, and South America.
The haplotype DRB1*0407/DQB1*0302/DPB1*0402 was found in 68.8% of Colombian patients (odds ratio 15.4; 95% confidence interval 6.7–35.9) and only in 12.5% of the controls. Zuloaga-Salcedo and colleagues proposed the existence of an HLA B39/DRB1*0407 haplotype, which includes class I and class II HLA alleles located in the sixth chromosome, as a susceptibility region for this disease in Mexican patients. Some possible protective alleles have also been described, including HLA DRB1*0802 in Mexicans and HLA DRB1*01 and DRB1*13 in Colombians.
Although AP is strongly associated with HLA DR4, the disease may occur in the absence of this HLA subtype, leading Grabczynska and colleagues to propose that there are other genes in the HLA DR region or its vicinity that may contribute to AP susceptibility. Some investigators proposed that AP is a persistent variant of polymorphic light eruption, in the presence of predisposing haplotypes and precipitation by environmental factors. This view is supported by reported cases of coexistence of polymorphic light eruption and AP, or progression of polymorphic light eruption to AP and vice versa. However, it should be noted that the combination of polymorphic light eruption, HLA DR4, and a family history of AP is not always associated with the development of AP. Therefore, Latin American dermatologists consider AP as a distinct entity.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


