Cutaneous CD4+ small–medium T-cell Lymphoma
Cutaneous CD4+ small–medium T-cell lymphoma is listed as a provisional entity in the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues [1]. In the past it has been described mostly under the term “cutaneous small/medium pleomorphic T-cell lymphoma.” Since the first description in 1995 by Friedmann et al. [2] the existence of this lymphoma type has been the subject of numerous debates and controversial interpretations, and probably a heterogeneous group of patients has been included in some studies [3–5].
Since mycosis fungoides is a cutaneous T-cell lymphoma characterized by the predominance of small–medium pleomorphic CD4+ T lymphocytes, the diagnosis of cutaneous CD4+ small–medium T-cell lymphoma can only be accepted if mycosis fungoides is excluded by a complete clinical examination. In fact, a careful re-examination of the clinical pictures of the first cases reported as cutaneous small–medium pleomorphic T-cell lymphoma suggests that at least some of them were actually examples of mycosis fungoides.
In spite of two decades of discussion since the first description, there is still no consensus on the existence, definition, and classification of cutaneous CD4+ small–medium T-cell lymphoma as a distinct entity. Cases similar on clinical and histopathologic grounds to those reported as cutaneous CD4+ small–medium T-cell lymphoma have been published under different diagnoses, including “idiopathic pseudo T-cell lymphoma,” “pseudolymphomatous folliculitis,” “cutaneous lymphoid hyperplasia,” “solitary lymphomatoid papule, nodule or tumor,” “primary cutaneous follicular helper T-cell lymphoma,” “indolent CD8+ lymphoid proliferation of the face,” “primary cutaneous peripheral T-cell lymphoma, unspecified,” and “monoclonal atypical T-cell hyperplasia,” among others [6–11]. In addition, cases of aggressive cutaneous cytotoxic lymphomas may show a small–medium pleomorphic cytomorphology (see Chapter 6) and cases of peripheral T-cell lymphoma, not otherwise specified (NOS), may be indistinguishable from CD4+ small–medium T-cell lymphoma as well (see Chapter 7), thus generating more disarray in an already confused field (see also the section on final considerations at the end of this chapter). As prognosis of patients with solitary lesions is almost invariably benign, the term “small–medium pleomorphic T-cell nodule of undetermined significance” has been proposed for these lesions [12, 13].
A relationship between cutaneous CD4+ small–medium T-cell lymphoma and T follicular helper (TFH) lymphocytes has been postulated [9, 14]. Whether cases showing this phenotype represent a variant of the disease or a distinct lymphoma entity is not clear yet (see the section on primary cutaneous TFH-cell lymphoma in this chapter). Another phenotypic variant of cutaneous CD4+ small–medium T-cell lymphoma is most likely represented by the so-called “indolent CD8+ lymphoid proliferation of the ear (face)”, a condition characterized by similar clinical and histopathologic features but by a T-cytotoxic rather than T-helper phenotype (see the section on indolent CD8+ lymphoid proliferation of the ear (face) in this chapter) [11, 15–17].
In the context of the controversies mentioned above, the diagnosis of cutaneous CD4+ small–medium T-cell lymphoma should probably be restricted to cases characterized by the following features:
- absence of other lesions and/or clinical history of mycosis fungoides;
- solitary lesion;
- negative staging;
- nodular or diffuse infiltrates of small–medium pleomorphic (monoclonal) T lymphocytes admixed with many reactive cells, where only a few large cells may be present;
- absence of marked epidermotropism of neoplastic cells;
- α/β phenotype (CD4+, rarely CD8+);
- absence of CD30 expression, or expression restricted to a small minority of the cells.
Cases characterized by multiple lesions are indistinguishable from cutaneous peripheral T-cell lymphoma, NOS, and probably should not be lumped together with those characterized by solitary tumors, as course and prognosis are different.
Clinical Features
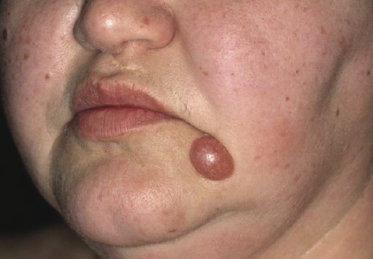
Patients are adults or elderly without a clear-cut gender predilection. Children may be affected rarely [18]. They present usually with solitary tumors, commonly located on the face and neck or upper trunk (Fig. 8.1). Patients with multiple tumors have been reported in the literature, but many of these cases may represent a different entity. The surface of the tumors is erythematous or purplish. Ulceration is uncommon.
Histopathology, Immunophenotype, and Molecular Genetics
Histopathology
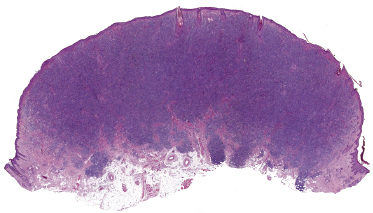
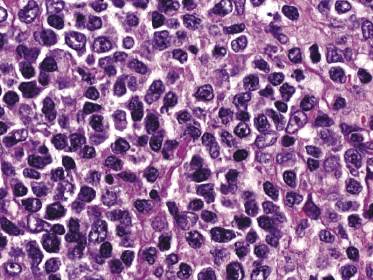
Histology reveals dense, nodular or diffuse lymphoid infiltrates within the entire dermis, often involving the superficial part of the subcutaneous fat (Fig. 8.2). Cytomorphology shows a predominance of small/medium-sized lymphocytes with pleomorphic nuclei (Fig. 8.3). Large cells, when present, should not exceed 30% of the neoplastic infiltrate [1, 19]. Epidermotropism is usually completely absent or present only focally. Many reactive cells are commonly found admixed with the neoplastic ones. A granulomatous reaction can be observed in a proportion of the cases and may sometimes be the cause of diagnostic problems [20]. Reactive germinal centers can be found, but are uncommon.
Immunophenotype
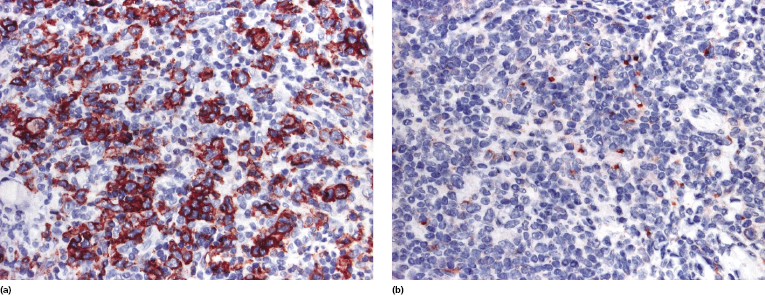
The neoplastic cells show a T-helper phenotype, sometimes with loss of pan-T-cell antigens (Fig. 8.4a to c). Staining for CD30 is negative or limited to a small minority of cells. A variably large, reactive infiltrate of B lymphocytes is commonly found (Fig. 8.4d). The proliferation rate is usually increased (Fig. 8.4e).
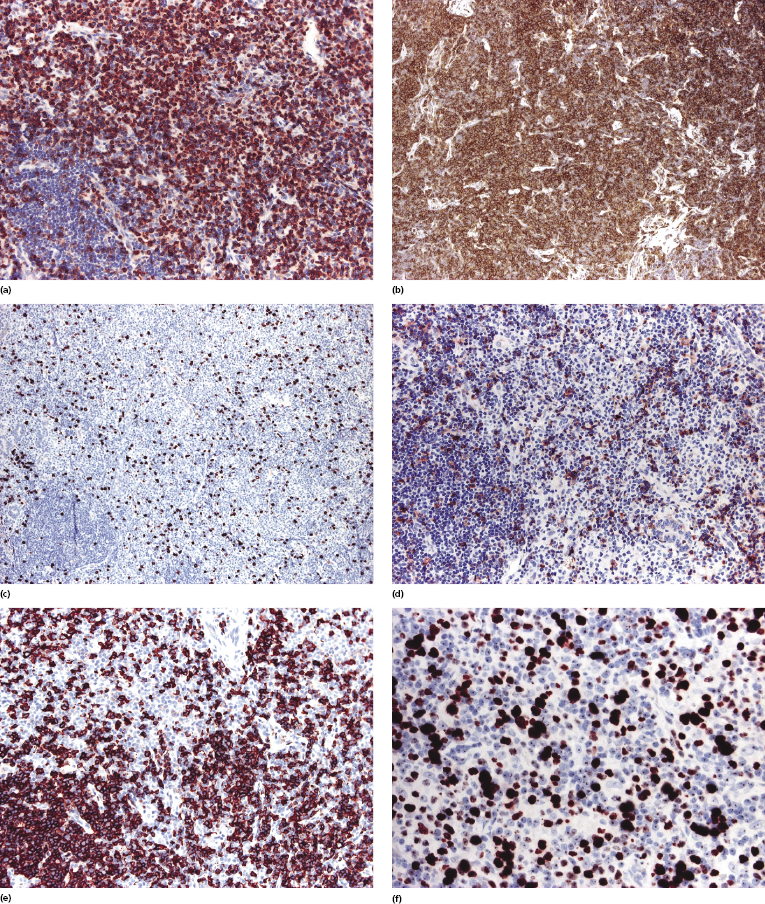
A cytotoxic, CD8+ variant of cutaneous CD4+ small–medium T-cell lymphoma is probably represented by so-called indolent CD8+ lymphoid proliferation of the face (see the section on indolent CD8+ lymphoid proliferation of the ear (face) in this chapter).
The expression of PD-1 by neoplastic cells of cutaneous CD4+ small–medium T-cell lymphoma has been described in some cases [9, 14] (Fig. 8.5). PD-1 expression can get lost in follow-up biopsies [21]. For a detailed discussion of such cases see the section on primary cutaneous T follicular helper (TFH)-cell lymphoma in this chapter.
Molecular Genetics
Molecular analysis of the T-cell receptor (TCR) genes rearrangement shows monoclonality of T lymphocytes in the majority of the cases. Specific genetic aberrations have not been identified.
The presence of Epstein–Barr virus (EBV) DNA has been detected by polymerase chain reaction (PCR) techniques in two cases of cutaneous CD4+ small–medium T-cell lymphoma [22], but this was most likely a false positive finding. The virus does not play any role in the pathogenesis of the disease.
Treatment
Most patients present with solitary tumors that can be treated by surgical excision alone, local radiotherapy, or a combination of the two. I have rarely seen patients showing spontaneous regression of the lesions after incisional biopsy.
In one study cyclophosphamide or interferon-α have been used for patients with generalized skin lesions [2]. It must be underlined that analysis of treatment modalities reported in the past is hindered by the inclusion of different entities in the various studies published. Multi-agent systemic chemotherapy should be reserved for cases with extracutaneous spread.
Prognosis
The potential of cutaneous CD4+ small–medium T-cell lymphoma for dissemination is unclear and evaluation of prognosis has been hindered by the difficulties in diagnosis and classification mentioned at the beginning of this chapter.
In the WHO classification an estimated 5-year survival of approximately 80% is reported [1], but it may be due to inclusion of more aggressive entities within this group of lymphomas. Patients with solitary lesions have an excellent prognosis [3, 12, 13, 18, 23–26]. On the other hand, cases that were associated with progression were almost invariably those showing multiple lesions at presentation [4]. Patients with lesions confined to the legs may have a more aggressive course (see Teaching case 8.1 in this chapter) [27].

Full access? Get Clinical Tree


