Sézary Syndrome
Sézary syndrome is characterized clinically by pruritic erythroderma, generalized lymphadenopathy, and the presence of circulating malignant T lymphocytes (Sézary cells). Other typical cutaneous changes include palmoplantar hyperkeratosis, alopecia, and onychodystrophy. In the World Health Organization (WHO) Classification of Tumours of Hematopoietic and Lymphoid Tissues, Sézary syndrome is considered as a specific type of T-cell lymphoma, clearly separated from mycosis fungoides [1]. In fact, there is a growing body of evidence showing that the two diseases represent distinct entities [2, 3].
Differentiation of Sézary syndrome from non-neoplastic erythroderma may be extremely difficult. The main causes of erythroderma, besides Sézary syndrome and mycosis fungoides, are atopic dermatitis, psoriasis, and drug reactions, but less frequently other cutaneous inflammatory disorders may present with erythrodermic features as well [4]. The differential diagnosis of erythroderma is considered one of the most vexing problems in dermatology and dermatopathology. In some cases the precise cause cannot be determined and only a descriptive diagnosis is used (“idiopathic erythroderma,” “homme rouge,” “red man syndrome”). For a thorough discussion on differential diagnosis of erythroderma see the section on non-neoplastic erythroderma in adult patients in Chapter 26. Erythrodermic mycosis fungoides (i.e., erythroderma developing in patients with a previous history of mycosis fungoides) should be distinguished from true Sézary syndrome (see Chapter 2) [1].
In the past, different diagnostic criteria have been used in studies on Sézary syndrome, hindering comparison of published data. In this context, the inclusion in many studies, even recent ones, of Sézary syndrome and mycosis fungoides in the same category of “cutaneous T-cell lymphoma” does not allow a clear interpretation of the data. The demonstration of the same monoclonal population of T lymphocytes within the peripheral blood and the skin is considered a crucial criterion for the diagnosis of Sézary syndrome [1, 5]. Other diagnostic criteria include the presence of at least 1000 circulating Sézary cells/mm3, an expanded CD4+ population in the peripheral blood resulting in a markedly increased CD4+:CD8+ ratio (>10), an increased population of CD4+/CD7− cells or CD4+/CD26− in the peripheral blood, and the loss of T-cell antigens such as CD2, CD3, CD4, and CD5 [1, 5]. As erythroderma may develop in different inflammatory conditions, and as neoplastic erythroderma may be observed in other cutaneous lymphomas such as mycosis fungoides and T-cell prolymphocytic leukemia, among others, it is important that strict criteria for diagnosis of Sézary syndrome are used. The presence of clonal T-cells within the peripheral blood alone should not prompt a diagnosis of Sézary syndrome. In fact, detection in the peripheral blood of a T-cell clone without clinical significance is not infrequent in elderly patients, and only the finding of identical clones in the blood and skin is considered diagnostic.
The use of standardized end points and response criteria is necessary, too, in order to compare the efficacy of various therapeutic agents on data collected at multiple sites and/or at different time points. The International Society for Cutaneous Lymphomas (ISCL), the United States Cutaneous Lymphoma Consortium (USCLC) and the European Organization for Research and Treatment of Cancer (EORTC)–Cutaneous Lymphoma Task Force have published a consensus statement on standardized end point and response criteria [6]. Complete response in the skin is defined as 100% clearance of skin lesions, partial response as 50–99% clearance without new tumors, stable disease as <25% increase to <50% clearance in skin disease without new tumors, and progressive disease as ≥25% increase in skin disease or new tumors [6]. Several end points have been defined, including the objective response rate, time to response, response duration, time to relapse/freedom from relapse, time to treatment failure/freedom from treatment failure, time to progression, progression-free survival, disease-free survival, and relapse-free survival [6].
A staging classification system for mycosis fungoides and Sézary syndrome was proposed by a joint working group of the ISCL and the EORTC–Cutaneous Lymphoma Task Force (see Chapter 2, Table 2.2) [7]. This system has been validated by a study on large numbers of patients [8]. Erythroderma is classified as T4 (corresponding to stages III and IV), and further stage determination is achieved according to the lymph nodes and blood status. Depending on the tumor burden in the peripheral blood, cases are classified as stage IIIB (B1: low blood tumor burden, >5% of peripheral blood lymphocytes are atypical cells but do not meet the criteria of B2; B1a: clone negative; B1b: clone positive) or IV (B2: high blood tumor burden, ≥1000/μL Sézary cells with positive clone). Patients with <5% of circulating Sézary cells are classified as B0 (stage III), and further subdivided into B0a (no monoclonality detected in the blood) and B0b (clonality detected).
A severity index that assigns different scores to various categories (body surface area, number of tumors, lymph node status, atypical cells in the peripheral blood, and visceral involvement) has been proposed as a tool for measuring disease activity [9]. Although numbers may be useful in assessing disease extent and activity, at the same time they convey a sense of precision that in some cases may be an oversimplification.
The etiology of Sézary syndrome is unknown. An association with viral infections has been postulated in the past, but ruled out in several studies. Although the disease may rarely develop in patients with long-standing inflammatory skin conditions, this association should be considered fortuitous and does not represent a link between Sézary syndrome and other cutaneous diseases. Sézary cells have a mature, memory phenotype with a helper T-cell type 2 (Th2) cytokine profile (characterized by expression of interleukin [IL]-4, IL-5, and IL-10). The vast majority of cases show high expression of the skin-homing T-cell chemokine receptor 4 (CCR4) [10]. As Sézary cells proliferate poorly, their accumulation in affected tissues may be due to defective T-cell homeostasis involving resistance to apoptosis rather than to increased cell division. Nonfunctional p53 has been detected in several cases [11], and one case with a complex p53 gene mutation has been observed in a Chernobyl survivor, suggesting a possible relationship with environmental factors [12]. Downregulation of FAS gene expression has been demonstrated in several studies, again pointing to a disturbance in the T-cell homeostasis [13–15]. T-plastin, associated with resistance to apoptosis, is expressed in blood lymphocytes of Sézary syndrome but not in those of mycosis fungoides or in healthy individuals, suggesting a role of this molecule in the pathogenesis of the disease [16, 17].
One of the main aspects of Sézary syndrome is the profound alteration of the immune system typical of the disease [18]. An increase in the tumor burden correlates with a decline in multiple arms of the cellular immune response, including natural killer (NK) and cytotoxic T lymphocytes implicated in antitumoral activity. NK cells and cytotoxic T lymphocytes may also be impaired by the production of IL-10 and transforming growth factor-β by the tumor cells. The number of myeloid dendritic cells and plasmacytoid dendritic cells is reduced in the peripheral blood, and lack of CD40 ligand expression by neoplastic cells may further contribute to the reduced antitumoral activity and impaired function of the immune system. The immune alterations are reversible upon successful treatment and normalization occurs if patients achieve complete remission. Due to the severe alterations of the immune system just mentioned, patients with Sézary syndrome have an increased risk of both serious infections and development of secondary malignancies [19].
Clinical Features
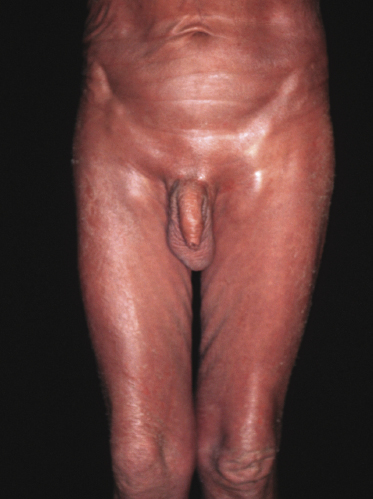
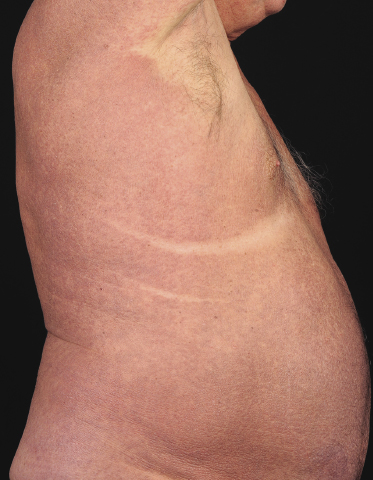
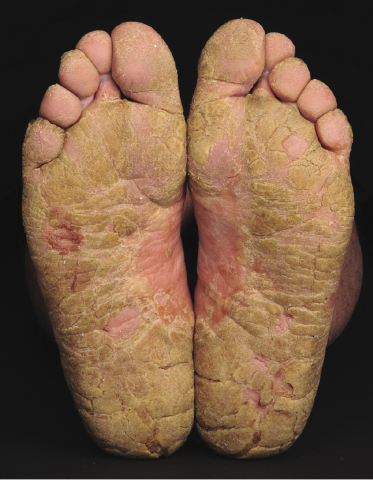
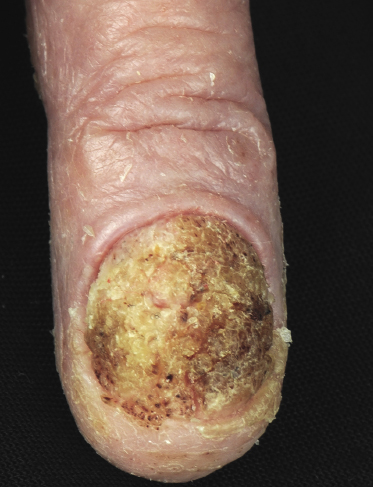
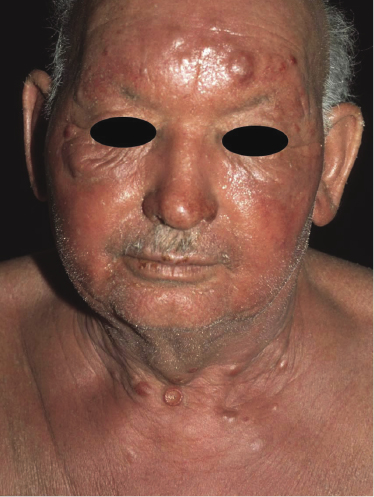
Sézary syndrome is a rare malignant T-cell lymphoma affecting elderly adults of both genders, with a predilection for males. Patients usually present with an abrupt onset of erythroderma and lymph node enlargement (Fig. 3.1), but itchy eczematous patches may be present for some time before erythroderma develops. Large skin folds (groins, axillae) may be spared (Fig. 3.2). The erythroderma is characterized by intense pruritus and scaling, and the pruritus may be intractable. Pruritus may be linked to production of IL-31 by neoplastic cells, and decreases when IL-31 levels are lower [19a]. Common clinical signs are the marked hyperkeratosis of the palms and soles, alopecia, and onychodystrophy (Figs 3.3 and 3.4). Ectropion may develop. As well as mycosis fungoides, Sézary syndrome may also be associated with follicular mucinosis [20, 21]. Other clinical variants include the presence of diffuse hyperpigmentation as a consequence of melanosis or hemosiderosis (melanoerythroderma) (Fig. 3.5) and of vesiculo-bullous lesions. In advanced stages tumors develop, usually on the background of erythroderma (Fig. 3.6).
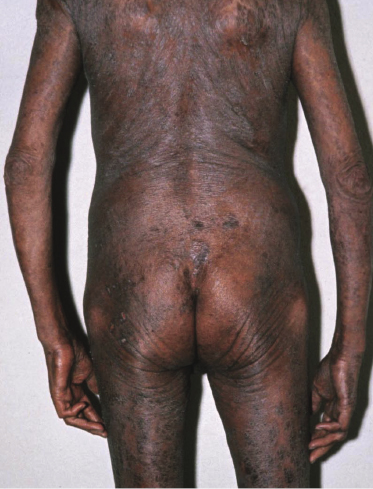
Peripheral lymph nodes are enlarged. Histopathologic analysis usually shows evidence of nodal involvement, but differentiation from so-called “dermopathic lymphadenopathy” may be very difficult. On the other hand, the presence of dermopathic lymphadenopathy is a bad prognostic sign even if a specific infiltrate cannot be demonstrated beyond doubt. Although Sézary syndrome is regarded as a leukemic variant of cutaneous T-cell lymphoma, involvement of the bone marrow is rare in the early phases (but it may be found in later stages).
Analysis of peripheral blood is necessary for the diagnosis of Sézary syndrome: diagnostic criteria include those mentioned above (presence of at least 1000 circulating Sézary cells/mm3, an expanded CD4+ population in the peripheral blood resulting in a markedly increased CD4+:CD8+ ratio (>10), an increased population of CD4+/CD7− cells or CD4+/CD26−, and the loss of T-cell antigens such as CD2, CD3, CD4, and CD5). However, CD7 and CD26 may be negative also in benign conditions. The demonstration of the same monoclonal population of T lymphocytes within the peripheral blood and the skin is a crucial diagnostic criterion. Other useful criteria include Sézary cells representing more than 20% of circulating lymphocytes, positivity for CD27 in the CD4+/CD26− T-cell population, presence of a CD158k+ clonal T-cell population, paucity of forkhead box protein P3 (FOX-P3)+ circulating cells, and presence of high-scatter T-cells [22, 23]. Flow cytometry analysis and analysis of T-cell receptor Vβ repertoires may be effective in demonstrating and quantifying small numbers of circulating tumor cells [24].
In some patients, prominent photosensitivity may mimic the picture of actinic reticuloid and be the source of a diagnostic pitfall. In fact, patients reported in the past as having actinic reticuloid with transformation into Sézary syndrome probably had Sézary syndrome from the outset (see also Chapter 26).
Histopathology, Immunophenotype, and Molecular Genetics
Histopathology
In general, the histopathologic aspects of skin lesions in Sézary syndrome are similar to those of mycosis fungoides [25–27], but epidermotropism is usually much less prominent [28]. Because of the lack of or minimal epidermotropism the histopathologic features are often difficult to interpret, and differentiation from benign erythroderma due to other diseases may be impossible [27]. When epidermotropism is present, typical Darier’s nests (Pautrier’s microabscesses) may be observed, indistinguishable from those seen in mycosis fungoides.
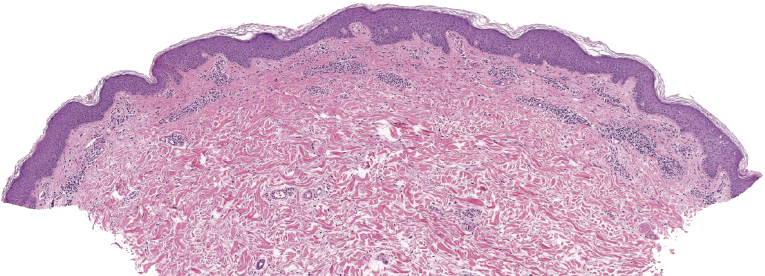
The epidermis shows in many cases as irregular or, less commonly, psoriasiform hyperplasia with variable degrees of spongiosis (Fig. 3.7). In most cases the infiltrate is only sparse and not clearly diagnostic of a cutaneous lymphoma (Fig. 3.8a and b). Although some atypical cells are usually present (Fig. 3.8c), they cannot be considered diagnostic of Sézary syndrome, as large, activated lymphocytes may be observed within the infiltrates of non-neoplastic erythroderma as well. Cytomorphology reveals predominance of small- to medium-sized pleomorphic (cerebriform) lymphocytes, often referred to as “Sézary cells” (Fig. 3.9). It must be underlined that cerebriform cells are observed only in dense infiltrates; thus for practical purposes, almost never in biopsies of early lesions, and in this context they cannot be considered to be a useful clue for early diagnosis. Because of the overlapping histopathological features, differential diagnosis from mycosis fungoides can be achieved only by correlation with the clinical picture.
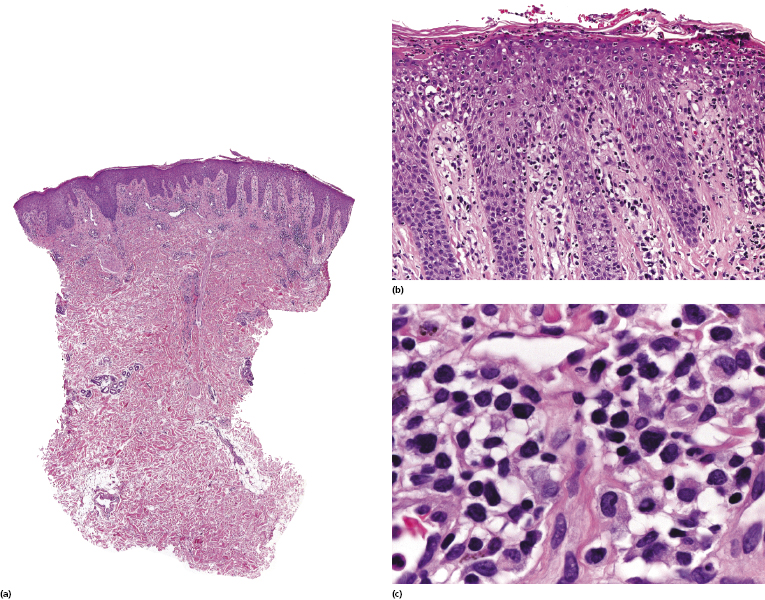
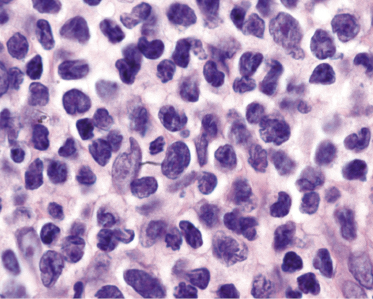
Histopathologic variants of Sézary syndrome include the presence of a prominent granulomatous reaction, deposition of mucin within hair follicles (follicular mucinosis), or large cell transformation [20, 21, 29]. Large cell transformation may be detected in skin lesions, lymph nodes, or both, and is indistinguishable from that occurring in advanced mycosis fungoides (see Chapter 2).
Immunophenotype
Immunohistology reveals a predominance of T-helper lymphocytes (CD3+, CD4+, CD7−, CD8−). The findings are indistinguishable from those observed in the majority of cases of mycosis fungoides. CD30 may be expressed in biopsies showing large cell transformation.
A different expression of programmed death-1 protein (PD-1, CD279) has been observed in neoplastic cells of Sézary syndrome (positive), as compared to those of mycosis fungoides (negative) [30, 31]. Some of the PD-1+ cases also express CD10, Bcl-6, and CXCL-13, thus showing a true T follicular helper (TFH) phenotype [32].
A subset of patients with Sézary syndrome shows a FOXP3+/CD25− phenotype of tumor cells, characterized in some cases by functional features of T-regulatory (Treg) cells [33–35]. The Treg phenotype has been associated with a worse prognosis in one study [33].
Molecular Genetics
In the majority of cases of Sézary syndrome, molecular genetic studies of skin lesions show a clonal rearrangement of the T-cell receptor (TCR) genes, but clonality may not be detected in biopsies of early stages. Once again, detection of the same clone within the peripheral blood and the skin is a crucial diagnostic criterion.
No specific karyotype is associated with Sézary syndrome, although genetic instability with recurrent chromosomal abnormalities are observed commonly. A panel of eight genes that can distinguish Sézary syndrome in patients with low numbers of circulating cells has been detected by the cDNA microarray technique [36]. Genome-wide analyses showed gains of 8q23–24.3 and 17q23–24, and losses of 9p21, 10p12–11.2, 10q22–24, 10q25–26, and 17p13–q11.1 [37]. Gains of 8q and 17q and losses of 10 and 17p have been observed also in other studies [38, 39] and could be detected even in cases with limited blood involvement and normal CD4+ cell count, confirming that these are common aberrations in Sézary syndrome. Losses on 10q23 affect the function of PTEN, a tumor suppressor gene involved in the regulation of the cell cycle, which is deleted or downregulated in most cases of Sézary syndrome [40]. In another study, copy number losses at 1p36p22, 6q24, and 15q11.2, as well as gains at 22q11.2q13.3, were capable of separating cases of Sézary syndrome from controls [41].
Downregulation of FAS and inactivation of p53 and p16 have been observed in several cases [13–15, 42]. BCL2 downregulation and JUNB upregulation may contribute to the resistance to apoptosis [43]. Using quantitative polymerase chain reaction (PCR) analyses, gain of cMYC and loss of cMYC antagonists (MXI1 and MNT) were observed in the majority of patients with Sézary syndrome, as well as alterations of the IL-2 pathway characterized by gains of STAT3/STAT5 and IL-2 receptor genes [44]. Derepression of MYC may be due also to loss of the transcription factor E2A encoded by the E2A gene, resulting in enhanced proliferation and cell cycle progression via activation of MYC and of the cell cycle regulator CDK6 [45]. Although conventional immunohistochemical stainings for CD158k did not allow separation of neoplastic from reactive cases, mRNA expression of CD158k/KIR3DR2 detected by real-time reverse transcription PCR was significantly higher in lesional skin of Sézary syndrome than in reactive erythrodermic conditions, suggesting that detection of CD158k/KIR3DR2 transcripts may represent a molecular tool for the diagnosis of Sézary syndrome [46].
Using RNA-Seq, an approach to transcriptome profiling that applies deep-sequencing technologies and can be used to obtain global cell transcriptomes, dysregulation of several key cancer pathways, including those involving phosphatidylinositol 3-kinase (PI3K), nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), and transforming growth factor β (TGFβ), have been observed in Sézary syndrome [47].
Studies focused on micro RNA (miRNA) expression profiling showed that miR-21 expression is upregulated and miR-342 is downregulated in Sézary syndrome, possibly playing a role in the regulation of apoptosis [48–51], while miR-214 and miR-199a are upregulated in Sézary syndrome but not in patients with erythrodermic atopic dermatitis, providing a possible differential diagnostic criterion [52].
Treatment
The therapy of Sézary syndrome remains unsatisfactory. The first-line treatment modalities include extracorporeal photopheresis, interferon-α, denileukin diftitox, low-dose methotrexate, and bexarotene. Comprehensive reviews of therapeutic options have been summarized by the EORTC and the USCLC [18, 53]. Guidelines for the use of extracorporeal photopheresis are also available [54]. Treatment modalities may be combined together or with psoralen + UV-A (PUVA). In the past, many patients were managed with chlorambucil combined with prednisone (Winkelmann scheme), but nowadays this is considered as a second-line therapeutic option.
As for mycosis fungoides, several other treatments have been applied in a limited number of patients with Sézary syndrome, particularly in therapy-refractory cases. The use of total skin electron beam is associated with improvement of the tumor burden in the peripheral blood [55–57]. Other modalities include multi-agent and single-agent chemotherapy, particularly gemcitabine, pentostatin [58, 59], histone deacetylase inhibitors like vorinostat or romidepsin [60], anti-CD52 antibody (alemtuzumab) [61], new retinoids such as trimetrexate [62], anti-IL-2 receptor antibody (daclizumab) [63], or liposomal doxorubicin [64]. Although all of these treatments show some degree of efficacy, at least in a proportion of patients, they do not change the natural history of the disease and recurrences are the rule. In addition, rapid progression of the disease and/or large cell transformation has been observed sometimes in patients treated with new therapeutic agents, particularly immunomodulatory drugs [65, 66].
In the last years, allogeneic stem cell transplantation preceded by reduced-intensity conditioning regimens has emerged as a potentially curative option, even in advanced stages of the disease [67–72]. A graft-versus-lymphoma mechanism may contribute to the beneficial effect of this treatment modality. Non-relapse mortality is lower in patients with good performance status treated with reduced-intensity conditioning regimens and with matched related donor [69].
New insights into the pathogenesis of Sézary syndrome may provide the rational basis for innovative treatment strategies in the future. As Notch 1 is overexpressed in tumor cells in a stage-dependent manner, Notch inhibition may represent a possible treatment [73, 74]. Indeed, Notch 1 downregulation induced apoptosis in cutaneous T-cell lymphoma cell lines [73]. Activation of peripheral blood dendritic cells by providing soluble recombinant CD40 ligand may also represent a novel strategy [75, 76]. Targeting IL-31 production may represent a promising way of treating pruritus in patients with Sézary syndrome [19a].
Prognosis
Overall survival of patients with Sézary syndrome depends on the diagnostic criteria used. The 5-year survival varies between 11% and almost 50% in published series, thus clearly showing that different criteria for diagnosis and classification were used in different centers. The disease-specific 5-year survival of 52 patients included in the Dutch and Austrian registries was 24% [77]. Even with improved therapeutic possibilities, the survival is still very poor [78]. In this context, it should be underlined that patients with erythroderma, even if the cause is not Sézary syndrome or mycosis fungoides (non-neoplastic erythroderma), have a decreased survival compared to healthy individuals [4].
If strict criteria are adopted, the 5-year survival of Sézary syndrome is poor [1, 77]. The prognostic value of the B1 and B2 categories of peripheral blood involvement according to the ISCL staging system was supported by one report [79], but a large study found a similar median survival for these two staging categories (3, 2 years and 3, 1 years, respectively) [8]. Patients with B0a category (<5% of Sézary cells in the peripheral blood and no T-cell clone) have a significantly better median survival than those with B0b (clone detected) (24, 5 vears and 6, 9 years, respectively) [8]. The presence of lymph nodes with dermopathic lymphadenitis (N1) is associated with a worse prognosis in comparison to N0 nodal status, confirming that, in spite of the lack of specific histopathologic criteria, dermopathic changes within the lymph nodes are a bad prognostic feature [8]. Patients with nodal stage Nx have a greater relative risk for death, underlining the need for excisional biopsies of palpable or radiologically enlarged lymph nodes [8]. The presence of an identical clone in the skin and peripheral blood is an independent prognostic criterion pointing to a worse prognosis [8, 80]. Other factors associated with reduced survival include the stage at diagnosis, failure to undergo remission after the first treatment, advanced age, male gender, race, elevated LDH and β2-microglobulin serum levels, presence of a high tumor burden in the peripheral blood, and prior exposure to multiple systemic drugs [81–85]. The expression of FOX-P3 by circulating Sézary cells has also been related to a worse prognosis [33]. The number of circulating Sézary cells may predict the response to treatment, with higher counts showing better responses.
A cutaneous lymphoma international prognostic index (CLIPi) has been proposed for assessing prognosis of patients with mycosis fungoides and Sézary syndrome [86]. Male gender, age >60, stages B1/B2 and N2/3, and visceral involvement were associated with worse prognosis. Three groups were stratified according to the number of prognostic factors: 0–1 factor (low risk), 2 factors (intermediate risk), and 3–5 factors (high risk).
Genetic data have also been used to predict prognosis in patients with Sézary syndrome. A panel of 10 genes that can identify a group of patients with survival shorter than 6 months has been identified by the cDNA microarray technique [36]. However, these results must be verified by larger studies. A shorter survival has been observed in patients with CDKN2A–CDKN2B inactivation [87]. Using reverse transcription-PCR, a group of 593 genes overexpressed in mycosis fungoides and Sézary syndrome were identified, allowing stratification of the disease in three different prognostic groups [88]. Genes involved in the Th17 pathway (e.g., IL-17F) were expressed in patients with a worse prognosis [88]. However, this study lumped together cases of advanced mycosis fungoides and Sézary syndrome under the term “cutaneous T-cell lymphoma,” thus hindering precise interpretation of the data.

Full access? Get Clinical Tree


