Other Cutaneous NK/T-Cell Lymphomas
Besides the entities discussed in the previous chapters, several other types of natural killer (NK)/T-cell lymphoma have been described at cutaneous sites, including cases arising both as primary and secondary in the skin. Clinicopathologic aspects of T-lymphoblastic lymphoma are summarized in Chapter 23, of T-prolymphocytic leukemia and aggressive NK-cell leukemia in Chapter 22, and of intravascular large NK/T-cell lymphoma in Chapter 14. Angioimmunoblastic T-cell lymphoma and hydroa vacciniforme-like lymphoma will be discussed in this chapter. Although hydroa vacciniforme-like lymphoma belongs to the group of cytotoxic NK/T-cell lymphomas, it has very peculiar and distinctive features, which are different from those of the three entities discussed in Chapter 6, and does not present major overlapping features with them.
Angioimmunoblastic T-Cell Lymphoma
Angioimmunoblastic T-cell lymphoma, formerly called angioimmunoblastic lymphadenopathy, is a peripheral T-cell lymphoma of follicular helper T lymphocytes (TFH) with a peculiar proliferation of high endothelial venules. It is recognized as a distinct entity in the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues [1]. Nonspecific skin manifestations have been described in several patients, including maculopapular eruptions, purpura, and erythroderma. Specific skin involvement is uncommon but it may be observed. Development of a second, Epstein–Barr virus (EBV)-associated B-cell lymphoma may occur in nodal cases.
Clinical Features, Histopathology, Immunophenotype, and Molecular Genetics
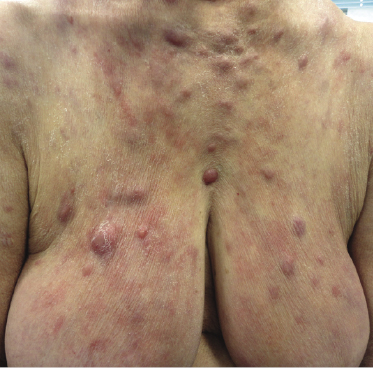
Patients are elderly adults, though cutaneous involvement in children has been reported [2]. Skin lesions may be the first manifestation of the disease [3, 4]. Specific cutaneous involvement in angioimmunoblastic T-cell lymphoma is characterized by nondescript erythematous papules, plaques, and tumors (Fig. 10.1).
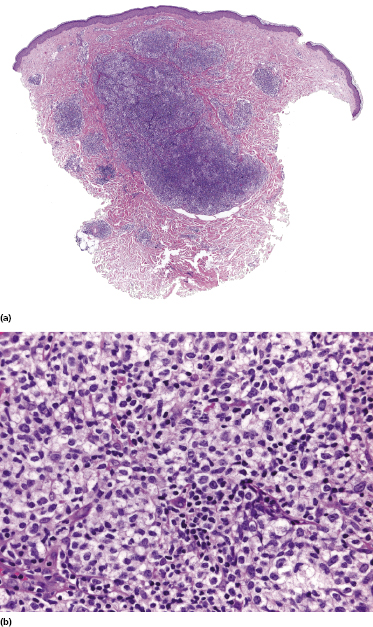
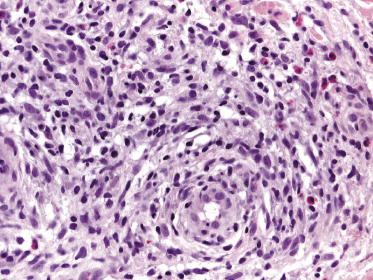
Histology shows nodular or rarely diffuse dermal infiltrates consisting of small-, medium-, or large-sized pleomorphic lymphocytes intermingled with reactive cells (plasma cells, eosinophils, histiocytes) (Fig. 10.2). The number of neoplastic lymphocytes is often a minority; thus the histopathologic features may be misinterpreted as those of a reactive infiltrate. Increased numbers of venules with a prominent endothelial lining are typically found (“high endothelial venules”) (Fig. 10.3) [5, 6]. A histopathologic presentation resembling an infectious process has also been reported [7].
In some cases, cutaneous biopsies show only superficial perivascular infiltrates with mild atypia, but the presence of monoclonal populations of T lymphocytes in the majority of these cases suggests that these lesions also represent specific manifestations of the disease.
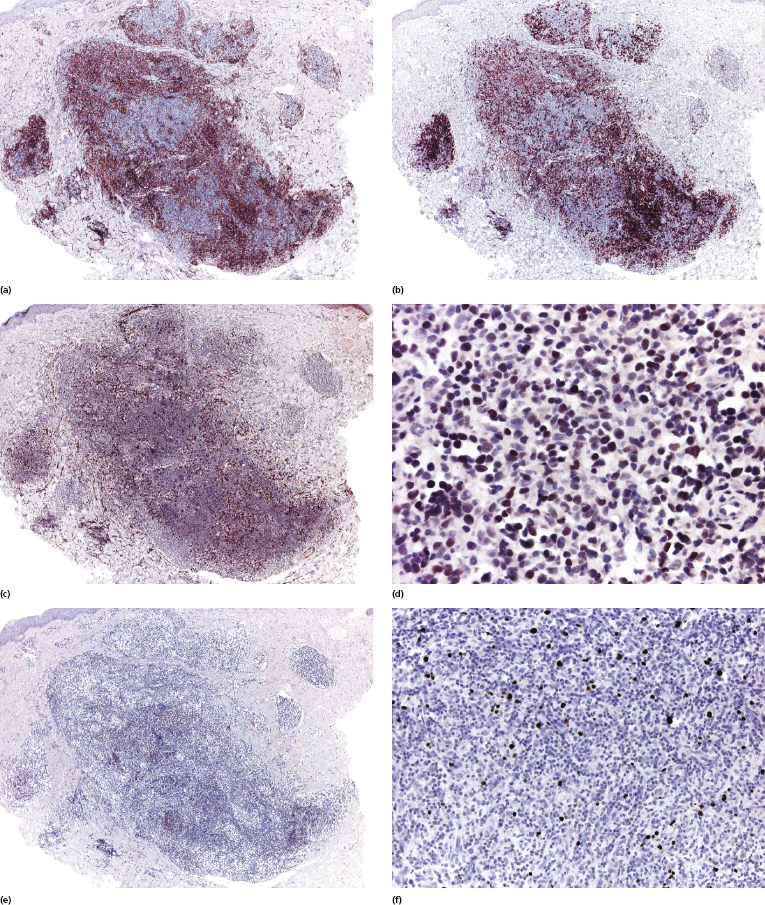
Neoplastic cells reveal a phenotype corresponding to TFH cells (CD3+, CD4+, CD8−, CD10+, Bcl-6+, PD-1+, chemokine ligand CXCL13+) (Fig. 10.4a to d). CXCL13+ is a chemokine which is expressed by helper T lymphocytes involved in the normal maturation process of germinal center B lymphocytes within the lymph nodes. Several studies have shown that CXCL13+ T lymphocytes can be identified in most cutaneous cases of angioimmunoblastic T-cell lymphoma, whereas expression of CD10 can be found in a minority of cases only [8–14]. In this context, I must underline that evaluation of CD10 in skin biopsies is very difficult because of strong background and positivity of dermal dendritic cells, and evaluation of a Bcl-6 staining is usually more informative (Fig. 10.4d). Clusters of polyclonal B lymphocytes are commonly present. Irregular aggregates of CD21+ follicular dendritic cells can be observed, especially around the high endothelial venules (Fig. 10.4e). EBV can be demonstrated within the B lymphocytes (Fig. 10.4f) [15], but the neoplastic T-cells are constantly negative. An increased number of TdT+ cells has been observed in lymph nodes of patients with angioimmunoblastic T-cell lymphoma [16].
Molecular genetics show a monoclonal rearrangement of T-cell receptor (TCR) genes and usually a polyclonal pattern of immunoglobulin (Ig) genes (a monoclonal rearrangement of the Ig genes can be found in 20–30% of cases). Trisomy 3 and 5 and an additional X chromosome are frequent genetic aberrations in nodal cases. Gains of 22q, 19, and 11q13 and losses of 13q have been shown in some cases by comparative genomic hybridization. In a case with cutaneous involvement, DNA microarrays in the affected lymph nodes revealed the expression of an apoptosis-inhibitory protein and of secondary lymphoid tissue chemokines, including tumor necrosis factor-β [17].
Treatment and Prognosis
There are only limited data on the prognosis and treatment of patients with specific skin involvement of angioimmunoblastic T-cell lymphoma. The prognosis is generally poor. Systemic treatment options include steroids, interferon-α, and chemotherapy. Chemotherapy may be difficult to administer due to frequent, potentially lethal infections. Onset of a second diffuse large B-cell lymphoma (usually, but not always, related to EBV) is a bad prognostic sign. Commencing therapy at an early stage of the disease may give better results in terms of survival.
Hydroa Vacciniforme-Like Lymphoma
A rare, peculiar NK/T-cell lymphoma affecting mainly children and resembling hydroa vacciniforme clinically has been described in Latin American (particularly Mexico, Guatemala, and Peru) and some Asiatic countries (China, Japan, Korea, and Taiwan) [18–28]. A putative case occurring in a Caucasian patient has also been reported [29]. Other terms used for this unusual lymphoproliferative disorder are hydroa-like lymphoma, hydroa vacciniforme-like T-cell lymphoma, atypical hydroa vacciniforme, angiocentric T-cell lymphoma of childhood, and edematous, scarring vasculitic panniculitis. In the WHO 2008 Classification of Tumours of Haematopoietic and Lymphoid Tissues, this lymphoma is included as a provisional entity in the group of EBV+ lymphoproliferative disorders of childhood [30].
The relationship of this disorder to conventional hydroa vacciniforme is unclear, but cases with intermediate features have been described (“borderline” hydroa vacciniforme/hydroa vacciniforme-like lymphoma) (see also Chapter 26) [31–34]. EBV is implicated in the etiology and pathogenesis of both disorders and it may be that hydroa vacciniforme-like lymphoma represents the malignant end of the spectrum of hydroa vacciniforme, perhaps arising only in genetically predisposed individuals.
Clinical Features, Histopathology, Immunophenotype, and Molecular Genetics
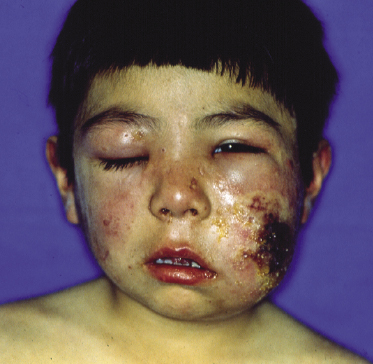
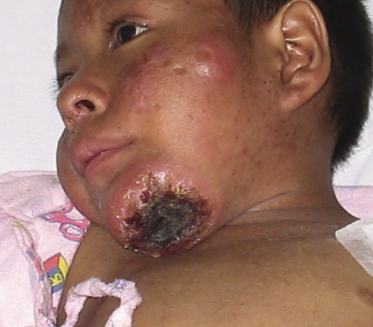
Most reported patients are children, but rarely adults with a similar clinical presentation have been observed. Lesions arise especially on sun-exposed areas, particularly the face and ears, the lower parts of the arms and the back of the hands, but other lesions are found on sun-covered skin, too. Facial swelling is common. Photosensitivity is invariably found, as well as hypersensitivity to mosquito bites. Clinically there are infiltrated plaques, often crusted and/or ulcerated, associated with blisters, edema, and varioliform scars (Fig. 10.5). Large tumors may be observed (Fig. 10.6). Systemic symptoms are present in the vast majority of the patients and include malaise, fever, weight loss, and often lymphadenopathy and hepatosplenomegaly. Laboratory investigations may reveal decreased levels of hemoglobin and a low hematocrit level, in addition to other alterations.
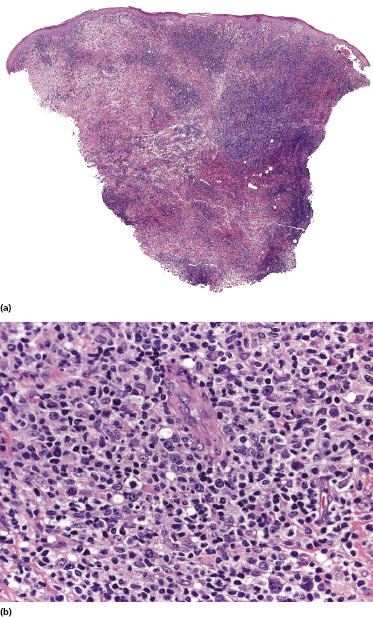
Histology is characterized by variably dense lymphoid infiltrates with atypical lymphocytes (atypia may be minimal in some cases, however), involving the entire dermis and in some cases the subcutaneous tissues (Fig. 10.7). Angiocentricity is frequently found, as well as infiltration of adnexal structures. The epidermis shows variable degrees of necrosis, spongiosis, and ulceration.
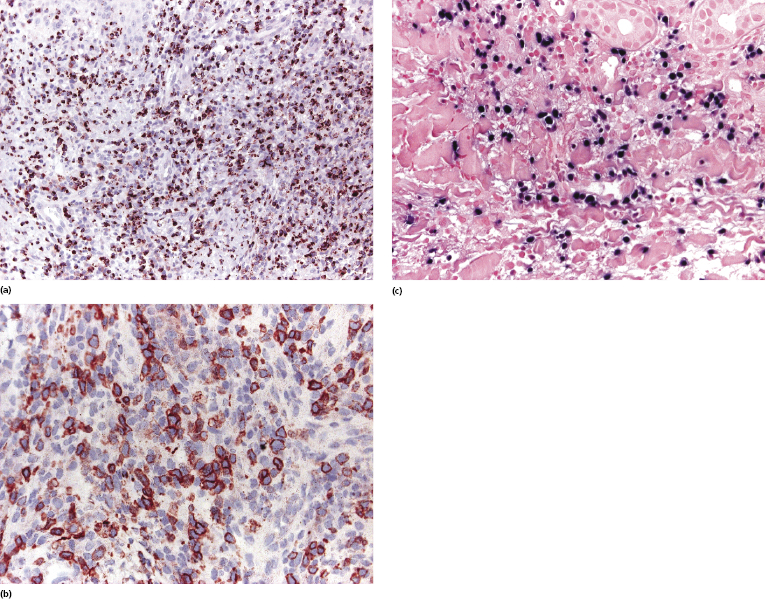
Neoplastic cells are positive for CD2 and CD3. Most cases reported expressed CD8 or were CD4−/CD8−, and were positive for cytotoxic proteins (TIA-1, granzyme B, perforin) (Fig. 10.8a). The αβ phenotype is much more common than the γδ or the true NK ones [35]. CD30 and CD56 expressions have been observed in a proportion of cases (Fig. 10.8b) [36, 37]. In situ hybridization for EBV shows a positive signal in variable numbers of tumor cells in practically all tested specimens (Fig. 10.8c). A monoclonal rearrangement of the TCR genes can be detected only in a minority of cases [29].
Treatment and Prognosis
The majority of patients with a diagnosis of hydroa vacciniforme-like lymphoma have been treated by systemic polychemotherapy, but it is not clear what the treatment of choice for “borderline” cases should be. Some patients have been managed with interferon-α with good responses.
Hydroa vacciniforme-like lymphoma has a prolonged course but a bad prognosis, intermediate between aggressive cutaneous cytotoxic NK/T-cell lymphomas and more indolent types of cutaneous T-cell lymphoma [26, 35]. Most cases published in the literature are either alive with specific manifestations of the disease or dead of systemic lymphoma. However, long-term follow-up data are missing and the published series are very small.

Full access? Get Clinical Tree


