1 Genetics, Prenatal Diagnosis and Counseling, and Feeding
Introduction
Cleft lip and/or palate formation is the most common congenital craniofacial abnormality, occurring in 1 in 500 to 1,000 live births. There are multiple genetic and environmental factors that have been previously associated with cleft lip and/or cleft palate (CP) formation.1 Families may or may not be aware of the diagnosis prenatally, and often are overwhelmed by the lifelong medical considerations that accompany the diagnosis. Patients born with cleft lip and/or CP will likely experience alterations in their swallowing, speech, hearing, dental occlusion, and outward appearance. Each of these alterations is determined, to a great degree, by the underlying origin of why the cleft lip and/or palate developed. To understand why cleft lip and/or palate occur, one must recall the early developmental processes active during normal lip and palate formation.
Embryology
Embryologic development occurs as the three embryonic layers form after fertilization takes place. The embryonic germ layers include the ectoderm (gives rise to the skin and nervous system), the endoderm (gives rise to the respiratory and gastrointestinal system), and the mesoderm (gives rise to the muscle and bone). Facial development begins as the neuropore is closing and the forebrain and midbrain are elongating. As the anterior neuropore closes, the ectoderm cells delaminate (migrate from the epithelium to form the mesenchyme) and invade the underlying mesodermal cells, forming the cranial neural crest (CNC). The CNC cells migrate from the dorsal neural tube anteriorly into the frontonasal process and paired branchial arches. There are multiple guidance signals within the mesoderm, overlying ectoderm, and underlying endoderm that, together, designate the CNC cells’ migration and differentiation. Multiple transcription factors, including the Hox gene family, are well known to control DNA transcription and translation within the craniofacial area, producing secreted growth factors. Growth factor signaling occurs between the endoderm, mesoderm, ectoderm, and CNC to specify the cell state and cell type in each area of the face. Interruption of any of these transcriptional or growth factor signals may lead to aberrant CNC migration or cell fate determination—either of which may lead to cleft lip and/or palate formation.2
Much of what we know today about facial development comes from elegant studies performed on chick and mouse models. The CNC migration into the frontonasal prominence (FNP) contributes to the formation of the forehead, nasal dorsum, median and lateral nasal prominences, premaxilla, and philtrum ( Fig. 1.1 ; Table 1.1 ). The chick model has been particularly rich when determining the cellular contribution leading to frontonasal development, as avian species have such varied beak formation. During anterior elongation of the frontal lobes, a very thin layer of ectoderm covers the underlying neural tissue. The CNC cells migrate into the FNP, under the guidance of the growth factors Sonic hedgehog and fibroblast growth factor 8. Interruption of FNP development can be associated with multiple genetic defects including midline or lateral facial clefting (e.g., Tessier atypical craniofacial clefts) that oftentimes are associated with underlying brain developmental abnormalities (e.g., holoprosencephaly).
Upper lip formation occurs in choreographed sequence with FNP development where the paired swellings of the first branchial arches, the maxillary prominences, merge together in the midline. Migration of the CNC from the dorsal neural tube into the maxillary prominence allows the lateral and medial nasal prominences to join with the premaxillary segment. Interruption of the CNC enlargement of the lateral, medial, or premaxillary segment will cause a cleft (or separation) between these structures, leading to a cleft of the lip. Additionally, if the lateral, medial, and premaxillary segments enlarge sufficiently to appose, but the epithelium fails to disappear, a cleft lip results as well. Multiple genes have been identified to affect lip formation and are discussed in the next section.
The orbicularis oris muscle, which is mesodermal in origin, migrates into the forming lip from the branchial arch, but not from the FNP. This migration pattern explains why patients with a bilateral cleft lip do not have muscle in the premaxillary/prolabial segment, which is derived from the FNP. Failure of the medial and lateral nasal segments to form also affects the shape of the lower lateral nasal alar cartilages, where the cartilage is depressed on the cleft side ( Table 1.2 ). The components of the lip on the cleft and noncleft side invariably retain all of the components of the dermal (white roll, dry and wet vermilion) and subdermal lip (orbicularis oris muscle fibers), making repair of the lip a matter of reconnecting the interrupted embryonic components.
Palate formation is a complex choreography of elongation, elevation, and fusion of paired maxillary prominences. Paired palate shelves initially begin to elongate from the maxilla on either side of the tongue. The developing mandible grows and pulls the tongue anterior and downward, allowing the palate shelves to elevate above the tongue. Once above the tongue, the paired palate shelves appose. Endodermal dissolution leads to palate shelf fusion. The palate shelves are comprised mostly of CNC cells in the anterior palate, giving rise to the hard palate, whereas the soft palate is comprised of muscle that is mesodermal in origin. Any interruption of palate shelf elongation, elevation, or fusion will lead to CP formation.

Week of Development | |
Facial primordia invaded by neural crest | 4th week |
Paired branchial arches develop | 7th week |
Medial and lateral nasal prominences fuse | 12th week |
A cleft, or separation, of the palate may present in a variety of phenotypes. The submucosal cleft palate is the most diminutive form, represented by some degree of dehiscence of the levator veli palatine muscles in the mid-line of the soft palate, bifid uvula, and possible notching the posterior hard palate. The severity of CP presentation continues from a cleft of the soft palate only to clefting through the soft and hard palates, which may be unilateral or bilateral.3 Multiple factors can interrupt palate elevation and fusion, and many of these are genetic. Therefore, all patients diagnosed with cleft lip and/or palate should be evaluated by a geneticist.
Relation | Risk of Recurrence |
Offspring of affected parent | 3% |
Sibling of affected person | 5% |
Sibling of two affected people | 10% |
Affected parent and sibling | 14% |
Genetics
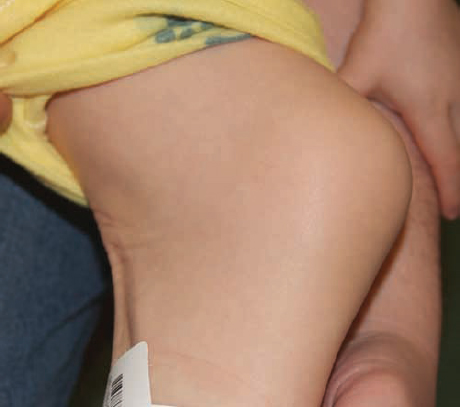
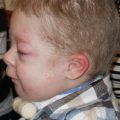
Cleft lip and/or palate occur in the setting of multiple genetic and environmental influences. There are over 400 genes that are linked to cleft lip and palate formation. Cleft lip and palate occurs in a syndromic fashion and with other associated physical anomalies in 30% of cases, which often occur with predictable Mendelian inheritance. However, the majority of cleft lip and/or palate patients (70%) present without other associated physical anomalies and with unpredictable Mendelian inheritance. The majority of patients with cleft lip and/or CP will have no other physical findings (nonsyndromic).4 The risk of cleft lip and/or palate in a nonsyndromic patient increases with the number of affected family members ( Table 1.3 ). Patients with other associated anomalies and a named syndrome often have an identifiable or genetic cause, such as popliteal pterygium syndrome ( Fig. 1.2 ). Cleft lip and/or cleft palate may occur in an autosomal dominant or autosomal recessive inheritance pattern ( Table 1.4 ).5
Prenatal Diagnosis
With the explosion of medical technology and increasing expectations from the general population for improved medical care, the diagnosis of fetal anomalies in utero is not only more common but it is frequently demanded by the expecting family. Physicians must now counsel expecting families regarding the advanced diagnostic ultrasonography and answer questions about how to interpret the findings in relation to the family′s beliefs and priorities.
Methods for Prenatal Diagnosis
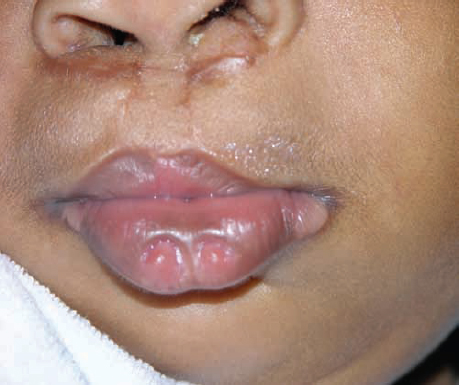
Today in the 21st century, prenatal ultrasound screening has become widely accepted for the diagnosis and documentation of the health of both mother and fetus. This has extended into developing countries. Routine two-dimensional (2D) ultrasounds are typically obtained during the first trimester to document viability. A second trimester ultrasound is performed with more emphasis on the development of the fetus ( Figs. 1.4 and 1.5 ).
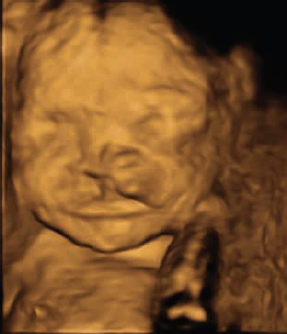
Since the 1970s there has been an increasing interest in improving the images of the fetal face using ultra-sound. The fetal face is frequently not imaged adequately in the first trimester screening ultrasounds because of fetal movement or obstruction of the view by the arms and hands. Three-dimensional (3D) ultrasound images of the face were first obtained in 1986. However, 3D ultrasound did not become widely used until the mid-1990s. By 2000, multiplanar volume rendering with 3D ultrasound became possible.6 The 2007 American Institute of Ultrasound in Medicine guidelines for prenatal ultrasound screening do not require visualization of the fetal face in the first trimester ultrasound; however, the fetal face must be imaged as a minimum requirement for the second trimester screening. The use of transvaginal ultrasound screening has improved the diagnostic accuracy, but it is still far from perfect.
Early studies revealed an improvement in prenatal cleft detection over time,1–5 presumably due to increased awareness and improved ultrasound technique. Overall, isolated CP proves more difficult to image than cleft lip +/− CP (CL+/−CP). In 2000, Stoll et al. reported that their detection rates using 2D ultrasound improved from 5.3 to 26.5% over a nearly 20-year period. It should be noted that all patients had a CL+/−CP. No instances of prenatal detection of an isolated CP were reported.7 Another study in 2000 reported a 17% detection rate, which increased if the cleft was associated with other anomalies.8
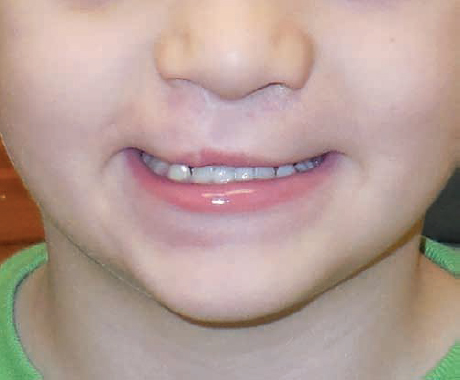
More recent studies report detection rates with 2D ultra-sound that vary widely by institutions worldwide. Detection rates for CL+/−CP range from 20 to 88%, and isolated CP detection ranged from 0 to 10%. Many of these studies note an improvement over time in their detection rates as experience and expertise in handling prenatal diagnosis improved.9–18 The limitations of these studies include a number of inaccuracies or false-positives that may be obtained. It is interesting to note that only four of the studies commented on inaccuracy rates, which ranged from 4 to 68%. Families should be counseled on potential inaccuracy in detecting a cleft and the possibility of a differing diagnosis at birth.9,10,13,15
Technological improvements, namely 3D and four-dimensional (4D) ultrasound, have improved accuracy, and the resulting image capture photograph is more recognizable to parents; however, diagnosing the isolated CP remains problematic.19 The ultrasound literature abounds with descriptions of different techniques for the 3D/4D imaging, which have infrequently successfully detected the isolated CP.20 In addition, with volume averaging, false-positives (a cleft is detected when not present) can appear due to shadowing.10
It has been suggested that magnetic resonance imaging (MRI) be considered for the diagnosis of secondary CP.21 Although this would assist the team in counseling the expectant parents, the cost–benefit analysis of such a screening technique is unsupported as of yet. The current recommendations for obtaining a fetal MRI generally center around detecting or further delineating anomalies that relate to the following: (1) infant prognosis, (2) change in maternal management, (3) to allow discussion of fetal surgery, or (4) planning the delivery timing. These are usually are not problematic in the infant with isolated CL+/−CP.22 The possible exception to this rule may be a fetus diagnosed with micrognathia. In the setting of suspected micrognathia, the severity of airway obstruction and the presence of associated anomalies are evaluated with 3D/4D ultrasound and amniocentesis to delineate the risk factors.23–25
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


