Fig. 6.1
(a) Clinical features of chronic plaque-type psoriasis. Clinical pictures of chronic plaque-type psoriasis characterized by scaly, red, extended skin lesions. In (b), note psoriatic lesion surrounding a scar
Guttate psoriasis is characterized by multiple small scaly plaques with often sudden onset, usually within few weeks after a bacterial infection of the upper airways, notably streptococcal pharyngitis in children and young adults [8].
GPP is a rare but potentially life-threatening disease characterized by episodic, widespread skin and systemic inflammation including high fever, fatigue, and neutrophil leukocytosis. Recent genetic data support the hypothesis that GPP is a disease of distinct etiology, being inherited as an autosomal recessive due with mutations in the IL36RN gene encoding the anti-inflammatory IL-36-receptor antagonist, IL-36Ra [9, 10].
Erythrodermic psoriasis is characterized by diffuse erythema, with or without scaling, and represents the most severe, albeit rare, phenotype.
About 20–30 % of psoriasis patients develop psoriatic arthritis (PsA), a seronegative, chronic, inflammatory musculoskeletal disorder with a wide spectrum of clinical disease presentation, expression, and clinical course [11, 12]. Since about 80 % of the patients develop PsA following psoriasis [13], PsA is sometimes considered as a disease within a disease, with the skin manifestation being the parent disease [14].
Psoriasis etiopathogenesis is that of a complex disease with disease initiation taking place in genetically predisposed individuals, in which a dysregulated immune response occurs following exposure to certain environmental triggers.
Genetic predisposition to psoriasis is supported by population and family studies as well as higher concordance rates in monozygotic twins, compared with dizygotic twins (up to 73 vs 20 %, depending on the population studied) [15–18]. Large efforts have been made in the past two decades to understand the genetic architecture of psoriasis. The psoriasis genetic landscape emerging from recent genome-wide association studies (GWAS) (See also Sect. 6.7) and their meta-analysis [19–28] includes 36 independent psoriasis-associated genetic regions in individuals of European ancestry, plus five more uniquely associated in the Chinese population [29]. Psoriasis susceptibility genes encompass skin-specific genes and immune-related genes, with the latter belonging to either the innate or the adaptive immunity, as well as bridging the two arms of the immune system. Psoriasis susceptibility region 1 (PSORS1) within the major histocompatibility complex is the strongest susceptibility locus [30, 31], and the HLA-Cw*0602 allele of the MHC class I molecule HLA-C is considered to be the primary associated allele, as confirmed by early sequence and haplotype analysis [32], genome-wide association studies (GWASs) [21, 22, 27], and analysis of high-density SNP data [33]. Among immune genes, the overrepresentation of four fundamental immunological processes and pathways strongly points towards their critical contribution to disease susceptibility: antigen presentation (HLA-C and ERAP1), NF-κb signaling (e.g., TNFAIP3, TNIP1, TRAF3IP2, CARD14), IL-23/IL-17 axis (e.g., IL-23, IL12B, and IL23R), and type I INF pathway (e.g., IL28RA and RNF114) [34]. The critical involvement in disease pathogenesis of the IL-23/IL17 pathway has been particularly well documented by a wealth of clinical and experimental studies showing a pivotal role for IL-23-induced and IL-17-mediated responses in psoriasis [35]. Moreover, the genetic association with IL23R is one of the very few supported by functional evidence with reduced IL-17 responses in carriers of the protective Arg381Gln IL23R allele [36, 37].
In contrast to the fast-growing list of psoriasis susceptibility genes, the environmental factors initiating the disease are still ill defined. Among known environmental triggers of psoriasis are drugs (the antiviral and antiproliferative agent imiquimod, lithium, beta-blockers, the cytokine INF-α, and anti-cytokine therapies such as anti-TNF agents), infections (streptococcal, HIV), physical trauma (tattoos, scars), smoking, alcohol, and stress [8].
The contribution of the immune system to psoriasis is not less complex than the overall disease pathogenesis, with a variety of innate and adaptive immune cells and proinflammatory mediators involved, possibly at different stages of the disease. The current view of psoriasis pathogenesis implies that the aberrant immune and epidermal response seen in psoriasis is sustained by a pathogenic cross talk between epithelial and immune cells [38, 39]. This interplay is primarily driven by the critical proinflammatory molecules, TNF, IL-23, and IL-17, whose direct therapeutic targeting has proven to be clinically effective, with other mediators, such as IFN-α, IFN-γ, and IL-22 also contributing to the initiation, amplification, and maintenance of the disease (Fig. 6.2).
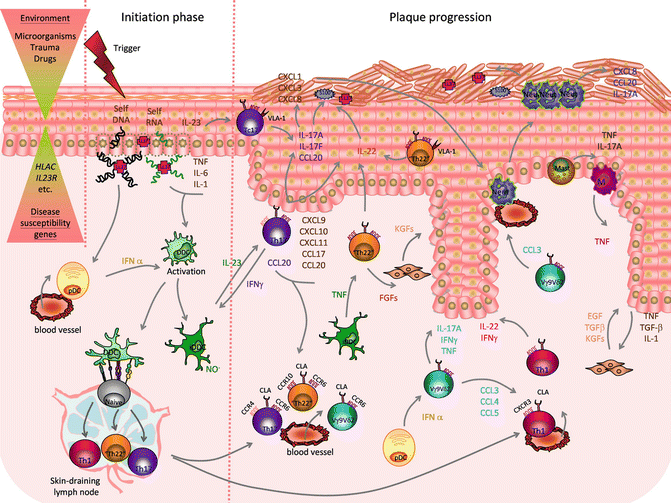
Fig. 6.2
Psoriasis etiopathogenesis. The combination of environmental factors with psoriasis susceptibility genes triggers a cascade of pathogenic events leading to disease initiation and plaque formation. In the initiation phase, proinflammatory cross talk between injured or stressed keratinocytes (KCs), releasing self-nucleic acids and LL-37, recruited plasmacytoid dendritic cells (pDCs), activated dermal DC (DDCs), and inflammatory DDC (iDDCs), producing IL-23, TNF, and nitric oxide radicals (NO .), promote the activation of skin-resident and newly recruited T cells that lead to plaque formation. IL-23 stimulates T helper 17 (Th17) and T cytotoxic 17 (Tc17) cells, expressing cutaneous leukocyte antigen (CLA), CCR6, and CCR4, plus very late antigen (VLA)-1 in the epidermis, to release IL-17A, IL-17 F, IL-22, and IFN-γ. IFN-γ further activates DDC. IL-17A and IL-17 F act on KCs promoting production of T cells and neutrophil-attracting chemokines (CXCL1,3,8-11;CCL17-20) and antimicrobial peptides (AMPs): S100 proteins and LL-37. CCL-20 favors the recruitment of more Th17 cells. IL-22, also produced by Th1 cells, expressing CXCR3 and skin-homing marker CLA, and “Th22”/“Tc22” cells, expressing CCR6, CCR10, and CLA, induces epidermal hyperplasia by impairing KC terminal differentiation. Recruited unconventional Vγ9vδ2 T cells, expressing CLA and CCR6, are activated by pDC-derived IFN-α and release further proinflammatory cytokines (IL-17A, IFNγ, TNF) as well as neutrophils (Neut) and Th-1-attracting chemokines (CCL3-5). Infiltrating Neut, mast cells, and macrophages (M) contribute to the proinflammatory environment producing cytokines (IL-17A, TNF), AMPs (S100 proteins, LL-37), and chemokines. Cross talk between keratinocytes producing IL-1, TNF and transforming growth factor beta (TGFβ), and fibroblasts, which in turn release keratinocyte growth factor (KGF), epidermal growth factor (EGF), and TGFβ, and possibly Th22 cells releasing fibroblast growth factor (FGF), contributes to tissue reorganization (Reproduced with permission from Di Meglio et al. [38])
In the initiation phase, LL-37 released by KCs, following physical trauma (Koebner phenomenon) or infection, binds to self-DNA/RNA fragments [40, 41], released by stressed or dying keratinocytes. LL-37/self-DNA complexes activate pDCs to produce IFNα [40], while self-RNA-LL-37 complexes, keratinocyte-derived IL-1β, IL-6, TNF, and pDC-derived IFNα activate DC. DC migrate to the skin-draining lymph nodes to present as yet unknown antigen (either of self or of microbial origin) to naive T cells. DDC activation and their interaction with T cells are central to plaque progression as it creates an IL-23/IL-17 inflammatory environment in which DC and macrophage-derived IL-23 promote an IL-17-rich proinflammatory environment sustained by Th17 cells [42, 43], Tc17 [44–47], γδ-T cells [48, 49], NCR+ group 3 innate lymphocytes (ILC3) [50–52], and possibly neutrophils and mast cells [53], producing IL-17A and IL-17 F, as well as IL-22 and IFN-γ. IL-17A and IL-17 F, sharing high structural and functional homology, activate keratinocytes to produce an array of molecules with chemoattractant properties, including neutrophil (CXCL1, CXCL2, CXCL5, CXCL8)- and T-cell (CCL20)-recruiting chemokines and AMP (LL37, S100A7/8/9/15) [54, 55]. Moreover, IL-22 produced by Th, Tc, and NCR+ ILC3 mediates most of the epidermal hyperplasia by impairing KC differentiation [56, 57]. Finally, a recent study has identified IL-9-producing Th cells in psoriatic lesions although their pathogenic relevance has not been established to date [58].
6.3 Immunosuppressant Therapy
At present, there is no definitive cure for psoriasis, and all the available treatments aim at inducing disease remission for the longest period of time. Treatments are chosen according to disease severity; however, all patients should be counseled for their psychosocial disability [59].
Most psoriasis patients (65 %) present with a mild form of disease which is usually treated with topical agents, with local anti-inflammatory and/or antiproliferative action.
However, moderate (25 %) and severe disease (8 %) cases require systemic treatment. Systemic treatment usually comes after unsuccessful topical strategies, in a two-tiered approach where systemic therapy is used as a second-line treatment of moderate to severe psoriasis. Traditional systemic therapies aim at general immunosuppression and include the use of cyclosporine and/or methotrexate (MTX).
6.3.1 Cyclosporine
Cyclosporine was observed to have clinical activity in psoriasis more than 30 years ago [60] and having gained FDA approval in 1997 has been extensively used since. Cyclosporine is a calcineurin inhibitor that, by interfering with IL-2 production, selectively inhibits T cells [61], thus acting on one of the key immune players in psoriasis immunopathogenesis. Other effects include the depletion of dermal and epidermal macrophages [62] as well as the inhibition of keratinocyte hyperproliferation [63] and expression of adhesion molecules [64]. The benefits of treatment with cyclosporine are rapidly seen, and the efficacy is dose dependent. However, improved efficacy obtained by using high doses (higher than 5 mg/kg/day) is counteracted by increased side effects. Although generally well tolerated, safety concerns are related to nephrotoxicity and neurotoxicity [65]. To minimize toxicity, a reduced dose of cyclosporine can be combined with topical and systemic treatments [66].
6.3.2 Methotrexate
Methotrexate is an effective first-line oral therapy, and it is considered the gold-standard comparator for new drugs such as biologics. Its efficacy in psoriasis was discovered in the 1950s, and it was officially approved for this indication in the early 1970s [67]. Methotrexate is an antifolate prodrug which is converted in its active form within the cells where it inhibits the enzymes involved in DNA synthesis. Such interference with DNA replication is most effective at high doses, and it is the basis of its antiproliferative effect, widely applied in the anticancer therapy. However, the mechanism of action of this drug in the context of psoriasis accounts for both antiproliferative and anti-inflammatory effects, which are seen at the low drug doses used in psoriasis treatment. Methotrexate (MTX) is effective in chronic plaque psoriasis not responding to conventional therapy but also in pustular psoriasis, psoriatic erythroderma, and psoriatic arthritis [68].
The limitations of the use of methotrexate, especially as a long-term treatment, are related to the development of toxicities such as myelosuppression, hepatotoxicity, and pulmonary damage [69].
There is no doubt that both cyclosporine and methotrexate are effective in treating psoriasis. A direct comparative study [70] showed that both drugs have comparable efficacy in treating psoriasis. The side effects associated with both drugs require careful monitoring of the patients during treatment and supported the development of more targeted immunological therapies which are now available as biologic therapies. However, conventional systemic therapies can be up to 20 times cheaper than biologics [71].
6.4 Biologic Therapy
In the last decade, the advancements in understanding psoriasis immunopathogenesis have been translated into better therapies. In particular, antibody- or fusion protein-based drugs, known as biologics, have been developed to target a specific immune receptor or cytokine. Biologics have proven to be an effective third-line therapy in moderate-to-severe psoriasis patients, unresponsive to non-biologic systemic agents. There are currently five biologics approved for the treatment of psoriasis, targeting either T cells or cytokines such as TNF or IL-12/IL-23 [8] (Table 6.1).
Table 6.1
Biologics approved for the treatment of psoriasis
Mechanism of action | Name | Molecular target | Phase | Biologic | Administration route | Company | References |
|---|---|---|---|---|---|---|---|
Anti-T cells | Alefacept | CD2 | Approved 2003 (US) | Human LFA-3/IgG1 fusion protein | IM or IV | Biogen | |
Anti-cytokine | Etanercept | TNF | Approved 2004 (US and EU) | Human TNF-R(p75)-lgG1 fusion protein | SC | Amgen | |
Infliximab | TNF | Approved 2006 (US and EU) | Mouse-human IgG1 chimeric monoclonal antibody | IV | Janssen Biotech | ||
Adalimumab | TNF | Approved 2007 (EU) 2008 (US) | Human IgG1 monoclonal antibody | SC | Abbott | ||
Ustekinumab | IL12p40 (IL-12, IL-23) | Approved 2009 (US and EU) | Human IgG1 monoclonal antibody | SC | Janssen Biotech |
6.4.1 Anti-T-Cell Therapy
The first biologics to be approved for psoriasis treatment were anti-T-cell therapies (alefacept and efalizumab), targeting T-cell adhesion or activation.
Alefacept, approved in 2003, is an LFA-3/IgG1 fusion protein which binds CD2 on T cells, thus blocking the interaction with antigen-presenting cells (APC) and inducing antibody-dependent cytotoxicity. The approval of this first biologic drug for treating psoriasis followed the successful completion of phase 3 clinical trials [72, 73] showing that 40 % of patients achieved a PASI 75 (75 % reduction of the PASI) response.
Efalizumab is a humanized monoclonal antibody targeting CD11a (alpha chain of LFA-1) therefore blocking the interaction of T cells with both APC and the blood vessels, inhibiting cutaneous infiltration. Efalizumab showed good efficacy in phase 3 clinical trials [85–91], but three cases of progressive multifocal leukoencephalopathy [92] caused the withdrawal from the market in 2009, highlighting the importance of carefully monitoring the long-term safety of immunomodulatory therapies.
Overall, notwithstanding good efficacy, anti-T-cell-targeted agents associate with general immunosuppression, and there are now available alternative biologic drugs with more targeted effects and therefore a better safety profile.
6.4.2 Anti-cytokine Therapies
The importance of a cytokine network mediating the cross talk between immune cells and keratinocytes to sustain the inflammatory loop in psoriasis has been further confirmed by the clinical success of targeting key cytokine pathways with specific biologic therapies. The first cytokine whose blockade showed therapeutic benefit in psoriasis was TNF. This was a serendipitous discovery made during the treatment with the anti-TNF drug infliximab of a patient affected by inflammatory bowel disease with concomitant psoriasis [74]. Currently there are three anti-TNF biologics approved for psoriasis each of them blocking TNF in a slightly different manner: infliximab and adalimumab are monoclonal antibodies against TNF, while etanercept is a human p75 TNF receptor fusion protein. TNF inhibitor efficacy has been shown in phase III clinical trials with up to 80 % of patients under treatment achieving PASI75 within 10–12 weeks of treatment [75–82]. Disease resolution is accompanied by normalization of KC differentiation and proliferation, downregulation of DC activation markers and downstream effector molecules, as well as reduction of Th17 responses [93, 94]. Interestingly downregulation of IL-17 pathway genes correlates with successful therapy response to etanercept [94, 95].
The second category of anti-cytokine biologics approved for psoriasis includes an antibody blocking the p40 subunit shared by IL-12 and IL-23 (ustekinumab) thus simultaneously blocking IL-12 and IL-23. Its efficacy is high, with 67 % of patients achieving PASI75 at 12 weeks of treatment [83]. A direct comparison of etanercept and ustekinumab showed that, despite both drugs achieved a PASI75 response in most patients, ustekinumab was clinically superior to etanercept as evaluated by the Physician Global Assessment with similar safety over a 12-week period [84]. Moreover, among patients who did not respond to etanercept, half of them achieved PASI75 after crossing over ustekinumab for 12 weeks.
The downside of the high efficacy rates of biologics is the potential of some serious adverse events, such as opportunistic infections and reactivation of latent tuberculosis [96]. However etanercept and ustekinumab have shown a good long-term safety profile in studies assessing safety up to 4- and 5-year treatment [75–77, 97, 98].
6.4.3 Emerging Biologics
New biologics have been developed so to specifically target either IL-17 or IL-23, and these molecules are currently tested for their safety and efficacy in clinical trials (Table 6.2).
Table 6.2
Emerging biologics and small molecules for the treatment of psoriasis
Type | Mechanism of action | Name | Molecular target | Phase | Biologic/Compound | Administration route | Company | References |
|---|---|---|---|---|---|---|---|---|
Biologics | Anti-cytokine | Tildrakizumab (MK-3222) | IL-23p19 | Phase III | Humanized IgG1 monoclonal antibody | SC | Merck | |
Guselkumab (CNTO 1959) | IL-23p19 | Phase II | Human IgG1 monoclonal antibody | SC | Janssen Biotech | |||
BI655066 | IL-23p19 | Phase II | Humanized IgG1 monoclonal antibody | SC | Boehringer Ingelheim | [103] | ||
Brodalumab (AMG 827) | IL-17R | Phase III | Human IgG2 monoclonal antibody | SC | Amgen | [104] | ||
Ixekizumab (LY2439821) | IL-17 | Phase III | Humanized IgG4 monoclonal antibody | SC | Eli Lilly | [105] | ||
Secukinumab (AIN457) | IL-17 | Phase III | Human IgG1 monoclonal antibody | SC or IV | Novartis | [106] | ||
Small molecule | PDE4 inhibitor | Apremilast (CC-10004) | PDE4 | Phase III | N/A | Oral | Celgene | |
JAK inhibitor | Tofacitinib (CP-690,550) | JAK1 and JAK3 | Phase II | N/A | Oral | Pfizer | ||
Tofacitinib (CP-690,550) | JAK1 and JAK3 | Phase II | N/A | Topical | Pfizer | [111] |
Blockage of IL-17 is presently investigated using monoclonal antibodies targeting either IL-17A (ixekizumab and secukinumab) or IL-17RA (brodalumab). In the latter case, the inhibition is not limited to IL-17A but covers also IL-17 F, IL17A/F, IL-25, and potentially IL-17C. All anti-IL17 biologics have shown striking efficacy in phase 2 clinical trials with more than 70 % of patients achieving PASI 75 and more than half achieving a notable PASI 90 [104–106].
Phase III clinical trials are ongoing to further confirm the safety and efficacy of these drugs while comparing them with approved biologics such as ustekinumab and etanercept. At the molecular level, IL-17 blockade showed a greater magnitude in regulating the expression of genes synergistically regulated by IL-17 and TNF-α as compared to TNF blockade [112].
There are three antibodies (BI655066, tildrakizumab, and guselkumab) specifically targeting the IL-23p19 subunit which are currently being tested in phase II [103], III [99], or have recently completed phase II [101] clinical trials, respectively. Preliminary data for guselkumab and tildrakizumab, presented at the American Academy of Dermatology meeting in 2013 and 2014, are very encouraging. Phase IIb data for guselkumab showed up to 81 % of patients receiving the highest dose achieving PASI75 response [102], while phase 3 data for tildrakizumab showed PASI75 response rates of 64–74 %, [100].
6.5 Emerging Small Molecules
The therapy revolution introduced by biologics in psoriasis is challenged by the high cost of about £10 k per patient/per year [113] by about 20–30 % of nonresponder patients and by the loss of response of some patients due to the development of antidrug antibodies which can decrease the drug bioavailability or induce side effects [114, 115]. Moreover, clinicians prefer to go for a safer option, at least for milder forms of disease. This has encouraged the exploration of small molecules as an alternative therapeutic option. Small molecules are low molecular weight compounds which target specific intracellular molecules involved in pivotal cellular signaling pathways [116]. Based on the importance of the cytokine-driven inflammatory pathways in psoriasis, the most promising small molecules currently under testing (Table 6.2) are targeting key cellular components in cytokine signaling such as Janus kinases (JAK) as well as enzymes involved in cytokine production. Among the JAK inhibitors tofacitinib, which specifically inhibits JAK1 and 3 and is approved for RA treatment, showed good efficacy in phase II trials of both oral and topical formulation of the drug [109, 111]. 66.7 % of patients treated with the highest dose of oral tofacitinib reached the end point PASI 75 at 12 weeks as compared to 2 % in the placebo-controlled group in a phase 2b study [110]. The other small molecule showing good results in phase II trials is apremilast, a phosphodiesterase 4 inhibitor that by inhibiting an enzyme involved in the breakdown of cAMP, suppresses the production of proinflammatory cytokines. Phase IIb clinical trials [107, 108] showed that the drug is safe and well tolerated. Efficacy as compared to placebo was significant with 41 % of patients achieving PASI75 at week 16 at the highest drug dose, advocating for additional clinical studies to test this small molecule.
Despite the potentially lower efficacy of small molecules as compared to biologics, the advantages are in the cheaper manufacturing process, the route of administration (oral or topical vs injectable), and a good safety profile in some cases. Thus, their use in mild to moderate form of psoriasis is foreseeable, although long-term safety data are required.
6.6 Concepts and Principles of Personalized Medicine: Patient Stratification and Biomarkers
The aspiration of tailoring medicine to the individual characteristics of each patient has long been a central vision of medicine. Pioneering studies resulting in the genetic diagnosis of Mendelian diseases and in the understanding that individual genetic variation affects how drugs are absorbed and metabolized laid the foundation of personalized medicine in the twentieth century.
In 1902, the first genetic disorder, alkaptonuria, was identified by Sir Archibald Garrod, who linked for the first time disease occurrence with genetic inheritance [117], laying the foundation of genetic diagnosis. In 1956, the genetic basis for selective drug toxicity was first postulated in the case of the antimalarial drug primaquine [118] and further strengthened later on by the discovery of the drug-metabolizing activity of the cytochrome P450 family enzymes [119] and the subsequent understanding of how their variation can affect the effective dose of a drug, launching the science of genetic variation in drug response or pharmacogenomics [120].
It is in the twenty-first century, however, that personalized medicine has begun in earnest, building upon the tremendous advances in the understanding of human diseases that have been enabled by the technological developments discussed in Sect. 6.7.
Genetics has taken the lead with a number of initiatives that have revolutionized our knowledge. The completion of the first draft of the human genome in 2001 [121, 122], complemented by the release and periodic update (1998–ongoing) of the Single Nucleotide Polymorphism Database (dbSNP, currently dbSNP 138) [123], provided the foundation for the advanced study of human genetics. Next, the launch and completion (2002–2009) of the International HapMap Project [124], listing allele frequencies and the correlation patterns between nearby gene variants, a phenomenon known as linkage disequilibrium (LD), across several populations for several million SNPs has made possible the discovery of disease susceptibility gene by means of genome-wide association studies (GWAS). Finally, the 1,000 Genome project (2008–ongoing) [125], aimed at sequencing the genome of 2,500 individuals from about 25 populations around the world, is poised to produce an extensive public catalog of human genetic variation to further support medical research studies, including personalized medicine approaches.
A number of objectives aimed at improving patient care are within the scope of personalized medicine: to predict individual susceptibility to disease, based on genetic or environmental factors; to detect the onset of disease at the very earliest stages; to predict and preempt disease progression; to develop novel targeted therapies; and to prescribe safe and effective medicines to each patient. The implementation of such objectives is also expected to increase the efficiency of the health-care system by improving quality, accessibility, and affordability.
In order to achieve such ambitious goals, personalized medicine is fast moving beyond the genome to encompass the entire spectrum of molecular medicine, including the proteome, metabolome, and epigenome, again fueled by improved knowledge and technology.
At the heart of personalized medicine is patient stratification, or the classification of individuals into subpopulations that differ in their susceptibility to a particular disease, or the natural history of their disease or their response to a specific treatment. Genetic and epigenetic testing, together with more conventional types of analysis (blood, urine, etc.), as well as with the careful consideration of lifestyle and other environmental factors, can effectively guide patient stratification, overcoming heterogeneity and aiding in disease diagnosis, prognosis, and therapy (Fig. 6.3).
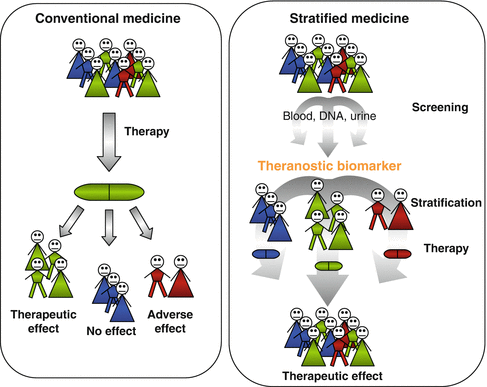
Fig. 6.3
Conventional versus stratified medicine. In conventional medicine approaches (left), patients receive the same drug which will have a therapeutic effect on the majority of them but will be ineffective in some and cause adverse events in others. In personalized medicine approaches, patients undergo screening using biological materials (DNA, urine, blood) to identify theranostic biomarkers allowing their stratification to receive the most appropriate and effective drug for each individual
Critical to the implementation of stratified and personalized medicine approaches are biomarkers or biological characteristics that are measured and evaluated objectively as an indicator of normal biological processes, pathogenic responses, or to pharmacological responses to therapeutic intervention.
Biomarkers can be classified in diagnostic biomarkers, indicating the existence of disease; prognostic biomarkers, able to forecast disease progression, with or without treatment; and predictive or theranostic biomarkers, able to predict the probable response to a particular treatment.
Thus, biomarkers can be distinguished in disease-related (diagnostic and prognostic) and drug-related biomarkers (pharmacokinetic and pharmacodynamic biomarker), the latter indicating how the patient’s body will process it and whether or not the drug will be effective.
An alternative classification, according to the NIH Biomarkers Definitions Working Group, distinguishes biomarkers in three categories: type 0 biomarkers, correlating longitudinally with the severity of disease; type 1 biomarkers, reflecting the effect of an intervention according to the mechanism of action of therapy itself (drug endotype); and type 2 biomarkers which are surrogate end points for a therapy [126].
Finally, biomarkers can be classified according to their ontogeny in genetic, epigenetics, transcriptional, soluble, and cellular biomarkers.
The classical biomarker discovery pipeline is a three-step process, from discovery to validation and clinical adoption [127]. A key element in translating biomarkers into clinical practice is the validation process. Tests used in the clinic to measure biomarkers must be reliable, with an acceptably low rate of false-positive and/or false-negative results. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) are key parameters to take in account when evaluating the performance of such tests [128]. Only biomarkers with high sensitivity and specificity can enter to the clinical practice, explaining why only a small fraction of potential biomarkers translate into clinical use.
Personalized medicine approaches are already established in modern clinical oncology, where a number of reliable biomarkers aiding in patients stratification have been identified and implemented in clinical practice. One of the first examples is the overexpression of the human epidermal growth factor receptor (Her2) in breast cancer tissue of certain patients. Her2 is a twofold biomarker, being a prognostic marker of more aggressive disease, but also a theranostic marker supporting the use of trastuzumab, a monoclonal antibody binding to Her2 used in the treatment of Her2-positive metastatic breast cancer [129].
6.7 Novel Technologies for Stratified Medicine in Psoriasis
The identification of biomarkers for patient stratification purposes can be done by using either a hypothesis- or a non-hypothesis-driven approach. While the former was the most common approach in the past, the latter has become a popular method in recent years thanks to the tremendous advancements in technologies in multiple areas of medical research. Overall, the recently developed technologies allow deep analysis of biological samples available in limited quantity, in a high-throughput manner permitting almost simultaneous genomics, proteomics, transcriptomics, and metabolomics analysis.
The most striking progresses have been made in the genomic sequencing field, where novel high-throughput technologies and next-generation sequencing (NGS) have enabled the genomics community to comprehensively characterize human DNA sequence variation, quantitating transcript abundance, detecting methylated region of the genome, and characterizing different gene isoforms. While a fully automated application of first-generation sequencing, such as the chain termination method developed by Sanger in 1975 [130], has been the mainstay for the original sequencing of the human genome, this has come with a cost of 3 billion dollars and took almost 10 years to complete [131]. The increasing demand for low-cost and high-throughput sequencing has driven the development of NGS technologies (second- and third-generation sequencing) that parallelize the sequencing process, producing thousands or millions of sequences concurrently, at a fraction of the initial costs. Moreover, the impact of these technologies extends far beyond genomic DNA sequencing. Traditional methods for studying DNA modification and its interactions with other cellular components or gene expression are being redesigned to take advantage of these powerful technologies, e.g., ChIP-Seq, to interrogate whole-genome histone modifications, whole-genome bisulfite sequencing of the DNA methylome, or deep-RNA sequencing [132, 133]. In psoriasis, high-throughput genotyping has enabled the identification of 36 psoriasis susceptibility genes by means of GWAS in which common genetic variations such as SNPs are examined in patients and control individuals to identify association with the disease [27]. Moreover, NSG have enabled the refining of the psoriasis transcriptome [134, 135] via RNA sequencing, building upon array-based analysis [136, 137].
Array-based technologies have also been used for DNA methylation profiling resulting in the identification of detection of different methylation profiles between lesional and nonlesional psoriatic skin [138].
Genetic, transcriptional, and epigenetic analysis can be complemented by the multiparameter analysis of samples at cellular level. Multidimensional analysis at single-cell level classically performed via flow cytometry has greatly improved in terms of number (up to 20 parameters) and types of markers (surface, intracellular, and phospho proteins) which can be measured thanks to advances in both instrumentations and reagents. Efforts have also been made to standardize the technology to be more applicable in clinical trials, for example, with the use of lyoplates, which are preformatted plates containing lyophilized cocktails of antibodies. Sample staining and acquisition for flow cytometry analysis using lypolates increase reproducibility, standardization, and medium throughput processing of the samples [139]. Intrinsic flow cytometry limitations in marker detection due to the spectral overlap of fluorochromes have been recently overcome by a novel technology for multiparameter cellular analysis named mass cytometry. By replacing the detection of fluorochromes with that of metal stable isotopes allows the investigation of up to 100 markers at the same time [140]. Studies using human samples have demonstrated the power of mass cytometry to identify highly diverse cell subsets [141, 142], resulting in a fine and detailed mapping of immune cells and their response. Moreover, mass cytometry-based analysis has recently elucidated a previously unappreciated role for CD8+ and γδ T cells in celiac disease following short-term gluten challenge, suggesting that immunological changes after short-term gluten exposure could be used to develop a novel diagnostic tool much faster than those currently in use requiring a longer exposure to the antigen [143].
Another powerful flow-based technology is phospho-flow cytometry, which quantifies the amount of phosphorylated intracellular signaling proteins before and after relevant cell stimulation, thus characterizing the functional state of complex immune cell populations in single individuals. Phospho-flow-based studies in immune-mediated disease such as SLE and RA have indicated signaling heterogeneity of different cell populations and different signaling profiles in different disease states [144], while similar studies are awaited in psoriasis.
Another area of recent investigation is metabolomics, which consists in the quantification of the metabolites in a biological system, by using complex technologies such as nuclear magnetic resonance and mass spectrometry. Both the quantification of specific metabolites and an untargeted and comprehensive metabolic profile associated to disease status can be obtained by metabolomics analysis [145]. This type of analysis has been proved successful in distinguishing different clinical phenotypes in complex disease such as CVD, cancer, and asthma [146]. Clinically relevant metabolite present in easily accessible samples such as urine could lead to the identification of metabolic biomarkers in psoriasis.
All the aforementioned technologies each generate a vast amount of data. Moreover, they are often used in combination to analyze the same biological sample, thus resulting in an escalating amount of data which require powerful bioinformatics tools not only to analyze, but also to integrate, handle, manage, and store these “omics” data [147]. Computational analysis approaches applied to the analysis of large data in psoriasis has already proven successful in a number of studies, especially taking advantage of the large amount of publicly available gene expression data from psoriasis skin. A meta-analytic approach has recently been used to combine the results of five microarray datasets obtaining the Meta-Analysis Derived (MAD) psoriasis transcriptome [134]. The overrepresentation of atherosclerosis signaling and fatty acid metabolism pathways in lesional skin support the close relationship between psoriasis and systemic manifestations [134]. A set of 20 “classifier” genes clearly separating lesional from nonlesional psoriasis skin has also been identified [134], to contain many genes that were part of the residual disease genomic profile, or “molecular scar’, still present in psoriasis skin after successful treatment [148] or genes with differential methylation status [138].
From skin gene expression data, ensembles of decision tree predictors were used to cluster psoriatic samples, and the analysis revealed distinct molecular subgroups within the clinical phenotype of plaque psoriasis [149]. In another study, cytokines and cell-type specific signatures were identified according to differentially expressed genes in the lesions, uncovering a range of inflammatory- and cytokine-associated gene expression patterns able to differentiate between etanercept responders and nonresponders [137].
Data from human samples can also be integrated with data from in vivo models to overcome the translational gap in the development of new targeted therapies. For example, the role of IL-22 in psoriasis has been recently evaluated by comparing the publicly available psoriasis transcriptome with the transcriptome derived from humanized mouse models of disease (i.e., IL-22 injection into xenografts of normal human skin) or inhibition of disease (i.e., antibody-mediated blockade of IL-22 into xenografts of psoriasis human skin). Mapping the in vivo experimental data over the psoriasis transcriptome led to the identification of the serine/threonine kinase PIM1, subsequently validated as a critical checkpoint for human skin inflammation and potential future therapeutic target in psoriasis [150]. Finally, a systems biology approach has been used to model and quantify immune cell interactions contributing to skin inflammation via cytokine signaling [151].
These new technologies provide high-throughput data platforms, shifting the analysis from discrete markers to the analysis of several interconnected systems. Such integrative approach is likely to result in the identification of novel biomarkers in the form of combination of markers or patterns instead of a single measurement.
6.8 Biomarkers in Stratified Medicine Approaches for Psoriasis
Large efforts aimed at the identification of psoriasis biomarkers are ongoing in order to implement stratified medicine approaches in disease management [127].
Psoriasis is diagnosed by clinical assessment by a dermatologist, with rare cases of histological confirmation of the disease. Thus, there is no urgent need for diagnostic biomarkers neither for disease monitoring during treatment as successful therapies are clinically evaluated by plaque resolution. On the contrary, prognostic biomarkers indicating disease severity progression or the onset of cardiovascular or other comorbidities would be extremely useful for better patient management. Moreover, biomarkers for early diagnosis are an unmet need in psoriasis arthritis, where the large disease heterogeneity often hampers proper diagnosis and the progressive course of the disease also calls prognostic biomarkers. Finally, despite the growing number of therapeutic alternatives to treat psoriasis, not all patients respond to the same treatment, and a recent survey has indicated that more than 50 % of patients polled are dissatisfied by the management of their disease [152]. The current therapeutic approach to treat psoriasis, especially in its moderate-to-severe forms, contemplates the use of different treatments, in an empirical attempt to find the most effective one. Patients are therefore likely to experience one or more ineffective therapies with relative associated side effects. Moreover, this approach also results in increased public health costs which are especially relevant in the case of expensive biologic drugs. Thus, biomarkers for better disease management are both a clinical and public health need which would benefit both patients and the health-care system. Despite large efforts in the quest for psoriasis biomarkers, none of those identified so far have entered into clinical use for disease diagnosis, prognosis, or for predicting therapy response [127]. Here, we describe them according to the type 0, 1, and 2 classification and discuss their potential use in stratified medicine approaches in psoriasis (Fig. 6.4).
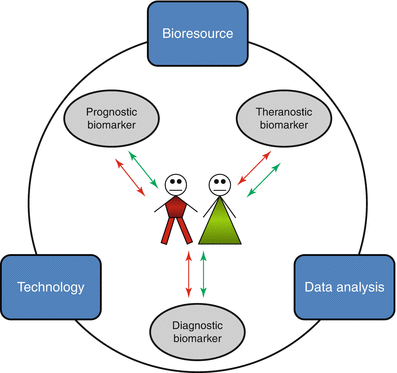
Fig. 6.4
Psoriasis biomarkers in translational research. Psoriasis patients would benefit from the clinical use of diagnostic, prognostic, and theranostic biomarkers. Translational research, involving the integrated use of bioresources, high-throughput technologies, and data analysis, aims to discover biomarkers for stratified medicine approaches in psoriasis
Related posts:
 Concept and Scientific Background of Personalized Medicine
Concept and Scientific Background of Personalized Medicine
 Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
 Melanoma: From Tumor-Specific Mutations to a New Molecular Taxonomy and Innovative Therapeutics
Melanoma: From Tumor-Specific Mutations to a New Molecular Taxonomy and Innovative Therapeutics
 Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
 Personalized Treatment in Cutaneous T-Cell Lymphoma (CTCL)
Personalized Treatment in Cutaneous T-Cell Lymphoma (CTCL)
 The Personalized Treatment for Urticaria
The Personalized Treatment for Urticaria
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


