5 Immunosuppression with the kidney as paradigm
Introduction
While the successful first kidney transplant was performed without immunosuppression, long-term graft survival after kidney transplantation requires maintenance immunosuppression except for those few fortunate situations of transplants between identical twins. Over the decades immunosuppressive regimens have become more sophisticated, allowing for longer-term graft survival and establishing kidney transplantation as the treatment modality of choice for patients with end-stage renal disease. Nevertheless immunosuppressive medications have significant toxicities and side-effects, and it has been the transplant community’s mission to find efficacious yet tolerable immunosuppressive combination regimens for transplant recipients. Undoubtedly the long-term goal would be to provide long-term graft acceptance without immunosuppression, but in clinical practice some degree of long-term immunosuppression is needed in most renal transplant recipients. In this chapter we will describe the commonly used immunosuppressive agents and how their combination has evolved in kidney transplantation. Figure 5.1 illustrates the mode of action of immunosuppressive drugs used in clinical practice.
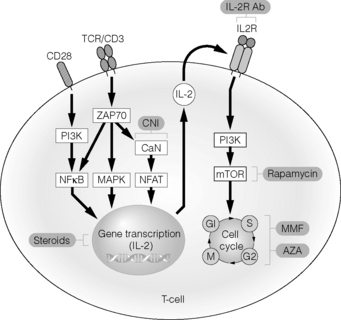
Figure 5.1 Schematic representation of mode of action of commonly used immunosuppressive drugs.
Reproduced from Demirkiran A, Hendrikx TK, Baan CC et al. Impact of immunosuppressive drugs on CD4+CD25+FOXP3+ regulatory T cells: does in vitro evidence translate to the clinical setting? Transplantation 2008; 85:783–9. With permission from Lippincott, Williams & Wilkins.
Azathioprine
Azathioprine (AZA), along with corticosteroids, was the earliest immunosuppressive medication used for the prevention of acute cellular rejection in kidney transplant recipients. Until the published reports by Murray et al. in 1963, total body or selective tissue irradiation was the primary method used to prevent rejection immediately after implantation of the transplanted kidney.1 This treatment resulted in unacceptable toxicity and was not effective long term. Transplant physicians began to employ pharmacological agents (AZA and corticosteroids) solely for the prevention of acute rejection.2 This resulted in acceptable graft survival rates and, after determining the appropriate doses of these medications, less toxicity.
AZA is a prodrug of 6-mercaptopurine (6-MP). Once absorbed into the bloodstream, AZA is completely converted to 6-MP. 6-MP exerts its immunosuppressive activity through purine synthesis inhibition. Purines (adenine, guanine and hypoxanthine) are necessary for DNA formation in new lymphocytes. 6-MP is transformed via hypoxanthine phosphoribosyltransferase (HGPRT) and a series of kinases into 6-thioguanine (6-TGN) nucleotides. It is then incorporated into elongating DNA chains, thus halting DNA chain elongation.3 By competing for HGPRT, 6-MP also renders this enzyme less available for true purines to be incorporated into DNA.
AZA is usually dosed at 1–3 mg/kg/day in a single daily dose, and undergoes multiple metabolic pathways. Once AZA is converted into 6-MP in the bloodstream, it may then be metabolised by three separate pathways: the thiopurine methyltransferase (TPMT) pathway, the HGPRT pathway or the xanthine oxidase (XO) pathway. Both the TPMT and XO pathways result in inactive metabolites. The HGPRT pathway results ultimately in the production of 6-TGN, which is incorporated into the elongating DNA chain or methylmercaptopurine nucleotide, which also inhibits de novo purine synthesis. The TPMT pathway is one of the best-described medication metabolic pathways subject to pharmacogenomic alterations. Approximately 10% of the population exhibits lower than normal TPMT activity, thereby forming fewer inactive metabolites and more active metabolites. These individuals tolerate only low doses of AZA. An important drug–drug interaction with AZA involves the coadministration of allopurinol, an XO inhibitor, usually used to prevent gouty arthritis attacks. By inhibiting XO, allopurinol eliminates or decreases one of 6-MP’s two metabolic pathways leading to inactive metabolites. This results in the formation of fewer inactive metabolites and more active metabolites. For patients who require treatment with allopurinol, substitution of AZA with an alternative immunosuppressive medication such as mycophenolate mofetil (MMF) is a possibility. The primary side-effect noted with AZA is bone marrow suppression.
In 1963, Murray et al. published the first case series using only medications for immunosuppression after kidney transplant.1 In 13 cadaveric and live-donor kidney transplant cases, they used AZA or 6-MP as their primary immunosuppressive agent. The first five patient cases had allograft function for less than 30 days, much of this due to AZA or 6-MP drug toxicity, which necessitated discontinuation of these medications. Discontinuation of these medications then resulted in rejection. Several patients also died due to complications from profound bone marrow suppression. In these early cases, AZA doses ranged from 6 to 12 mg/kg. The researchers quickly recognised that these doses resulted in profound toxicity. For the eight subsequent patients, doses were reduced to 2–4 mg/kg daily. Of these eight patients, at the time of publication, five had functioning grafts. The duration of these functioning grafts ranged from 40 to 365 days.
Shortly after Murray et al. published their results, Starzl and colleagues from the University of Colorado reported their experiences with an AZA-based immunosuppressive regimen.2 They published the results of 45 live-donor kidney transplant recipients, 32 of which received kidneys from related donors. These patients were treated with 2–4 mg/kg/day of AZA for 7–10 days prior to their kidney transplant, and daily thereafter. Doses were adjusted based on white blood cell indices. At the time of publication, 27 of the 45 transplanted patients were alive with functioning grafts. In these early days of transplantation, the balance between prevention and treatment of rejection and infectious complications was not well established. This was the cause of most post-transplant mortality. Despite the high rejection and mortality rates seen in these early case series, they represented a major milestone in the possibility of successful long-term kidney transplantation. These published series lead to the routine use of AZA as a primary maintenance immunosuppressive agent in kidney transplantation between the mid-1960s and the 1980s, when ciclosporin was studied.
The introduction of ciclosporin
After initial studies in healthy volunteers,4,5 followed by a pilot study in 18 renal transplant recipients,6 the new modified CYA was studied in 55 stable renal transplant recipients.7 The purpose of this study was to determine if trough CYA concentrations better predicted systemic exposure (area under the curve, AUC) with CYA microemulsion compared with non-modified CYA. The other purpose of the study was to determine if intraindividual variability was lower with the CYA microemulsion formulation compared with the non-modified formulation when studied in a larger stable kidney transplant population. This pharmacokinetic study included four study periods with two crossover time points. This analysis showed an increase of 59% and 30% in maximum concentration (Cmax) and AUC respectively with CYA microemulsion, when compared with non-modified CYA. There was also stronger correlation between CYA trough concentrations and AUC with the microemulsion (r2 = 0.806) compared with non-modified product (r2 = 0.492). In addition, there was less intrapatient variability for every measured pharmacokinetic parameter (tmax, Cmax, AUC and peak-to-trough fluctuation percentage, PTF%), except for trough concentrations. Although the intrapatient coefficient of variation (CV) between CYA microemulsion and non-modified CYA for trough concentrations was not statistically different, there was a strong trend towards decreased variability in this parameter as well (CV CYA microemulsion 14% vs. CV non-modified CYA 20%, P = 0.069).
Following this initial crossover study in renal transplant patients, a longer-term double-blind study was performed to compare pharmacokinetics between patients on non-modified and microemulsion CYA.8 This study included short-term (8 and 12 weeks) and longer-term (1 year) analyses. Fifty-nine stable renal transplant patients were randomised in a 1:4 fashion to either remain on their twice daily non-modified CYA regimen (n = 12) or switch (1 mg:1 mg) to CYA microemulsion (n = 47). A baseline pharmacokinetic evaluation was performed 1 week prior to randomisation, then again at weeks 8 and 12. For 46 patients who remained in the study for long-term analysis, a pharmacokinetic evaluation was performed again at 1 year. Similarly to the previous study, this study showed better correlation between trough concentrations and AUC with the microemulsion formulation (baseline/non-modified CYA: r2 = 0.507; CYA microemulsion week 8: r2 = 0.748; CYA microemulsion week 12: r2 = 0.822; CYA microemulsion 1 year: r2 = 0.576). Also, less intraindividual variability was shown with the microemulsion product in all pharmacokinetic parameters (tmax, Cmax, AUC and PTF%, P < 0.001), except trough concentrations.
Non-modified CYA was originally studied in two large randomised controlled trials published in the early 1980s. One study was conducted in Europe and the other in Canada.9,10 Both studies enrolled between 200 and 300 patients receiving cadaveric kidney transplants. The European study compared single-agent CYA treatment to AZA and corticosteroids. The Canadian study compared CYA and corticosteroids to best local therapy, which primarily consisted of AZA and prednisone. Some centres, however, did use antilymphocyte antibodies as part of their induction immunosuppression regimen in the control group. The European study at 1 and 5 years showed improved graft survival in the CYA-treated patients.11,12 At 1 year, graft survival was 72% in the CYA group and 52% in the AZA group (P = 0.001). This benefit was maintained at 5 years with graft survival of 55% in the CYA group and 40% in the AZA/prednisone group (P < 0.01). Patient survival was no different between the groups at either time point in the European study. The Canadian study showed both improved patient and graft survival in the CYA group compared to the AZA group at 3 years post-kidney transplant.13 Graft survival was 69% in the CYA group and 58% in the AZA group (P = 0.05), and patient survival was 90% in the CYA group and 82% in the AZA group (P = 0.04). This improvement in short- and intermediate-term graft survival with a new immunosuppressive medication would later prove to be one of the most significant contributions to kidney transplantation to date.
The introduction of tacrolimus and mycophenolate
After the introduction of CYA in the early 1980s, there were no new maintenance immunosuppressive medications approved for a little over 10 years. The mid-1990s, however, saw two new maintenance immunosuppressive medications that brought about substantial improvements in acute rejection rates in renal transplantation. Tacrolimus (TAC) and mycophenolate mofetil (MMF) would both prove to decrease acute rejection rates further when compared with traditional CYA-based immunosuppression. This added two new medications for transplant clinicians to use in maintenance immunosuppressive regimens.
TAC, the second CNI approved for clinical use, was approved in 1994 for the prevention of acute allograft rejection in liver transplant patients. TAC inhibits calcineurin by binding to FK binding protein 12 (FKBP12). This TAC–FKBP12 complex then binds with calcineurin, inhibiting the phosphatase activity of calcineurin. The inhibition of calcineurin further inhibits the dephosphorylation and translocation of NFAT. This ultimately inhibits the production of key cytokines involved in cell-mediated immune function, such as IL-2 and interferon-γ.14 TAC is usually administered orally every 12 hours. Its oral bioavailability ranges from 17% to 22%.14 Therapeutic drug monitoring for TAC is usually accomplished through 12-hour serum trough concentration monitoring, similar to CYA. Stated therapeutic doses for TAC range from 5 to 15 ng/mL, but depend largely upon patient-specific factors and concomitant immunosuppressive therapy. TAC is primarily metabolised by hepatic and intestinal cytochrome P450 3A4, and is subject to many drug interactions with other medications that either inhibit or induce this microenzyme.14 It is also a substrate of P glycoprotein and subject to drug interactions via this enzyme as well. Any addition or discontinuation of other medications that inhibit or induce these enzyme systems must be followed by TAC dosage adjustments and more intensive serum concentration monitoring.
TAC was first studied in the liver transplant population as part of a dual-drug regimen along with corticosteroids, and shown to be superior to CYA at preventing episodes of acute rejection. Two randomised open-label multicentre trials conducted in the early 1990s compared TAC/corticosteroid immunosuppression with CYA/AZA/corticosteroid immunosuppression in liver transplant recipients. Both the US and the European multicentre randomised trials concluded that at 1 year following a first-time liver transplant, TAC resulted in fewer episodes of acute rejection and refractory rejection.15,16
Shortly after being studied in liver transplant patients, TAC was studied in large numbers of kidney transplant patients, with similar results. Two similarly designed multicentre randomised, open-label, clinical trials were conducted in the USA and Europe.17,18 Each were large studies, with the US study randomising 412 patients and the European study 448 patients. Each of these trials compared regimens of TAC/AZA/corticosteroids to CYA (non-modified)/AZA/corticosteroids. The US study investigators used antibody induction therapy in all patients in addition to the above-described maintenance regimens.17 The primary outcome in each study was incidence of biopsy-proven acute rejection (BPAR) 1 year after cadaveric renal transplantation. Target CYA and TAC concentrations were similar in each study, with the US study targeting slightly higher CYA concentrations than the European study. Both studies targeted TAC troughs between 10 and 25 ng/mL for the first 3 months, then 5–15 ng/mL thereafter. CYA target concentrations in the two studies ranged between 100 and 400 for the first three months and 100–300 thereafter. Although open label in design, each study’s pathologists were blinded to treatment assignment to minimise bias in determination of rejection presence.
These two multicentre randomised trials in kidney transplantation showed similar positive results as had the earlier liver transplant studies. The US investigators reported BPAR rates of 30.7% in the TAC group and 46.4% in the CYA group (P = 0.001).17 The European investigators reported this same outcome in 25.9% of patients in the TAC arm and 45.7% of patients in the CYA arm (P < 0.001).18 At 1 year, there was no difference in either trial between groups for either patient survival or graft survival.
A 5-year follow-up of the US multicentre trial showed that improved outcomes with TAC proved durable.19 Although the intent-to-treat analysis failed to show any difference in patient or graft survival at 5 years, crossover analyses revealed improved outcomes in patients who were receiving TAC at year 5. Throughout the 5-year follow-up, 27.5% of patients crossed over from CYA to TAC, while only 9.3% crossed over from TAC to CYA (P < 0.001). The majority of these crossovers occurred in the first year of treatment; therefore the crossover analyses should represent long-term treatment on each medication. The majority (68.4%) of CYA randomised patients crossed over to TAC due to refractory rejection. Treatment failure, defined as graft loss or study drug discontinuation, was higher in the CYA group (56.3%) compared with the TAC group (43.8%, P = 0.008). In addition, graft survival was higher in African-Americans receiving TAC (65.4%) compared with CYA (42.6%, P = 0.013). Primary side-effects noted in most of the clinical trials are tremor, elevated blood glucose, nephrotoxicity and hypertension.
One possible weakness of the above studies was that the non-modified formulation of CYA was used. As previously discussed, CYA microemulsion demonstrates more reliable pharmacokinetics, which likely translates to better post-transplant outcomes. Since publication of these original comparisons between CYA (non-modified) and TAC in kidney transplants, comparison between TAC and CYA microemulsion has also been conducted. Johnson et al. studied three different immunosuppressive regimens in patients receiving a first cadaveric kidney transplant.20 They randomised 223 patients to open-label treatment with either CYA/MMF (n = 75), TAC/MMF (n = 72) or TAC/AZA (n = 76). Target trough concentrations for CYA and TAC were similar to the previously discussed studies. Primary end-points were again patient and graft survival, BPAR, development of insulin dependence, and renal function.
The primary outcomes of this 1-year analysis remained the same in a 3-year follow-up analysis.21 The authors additionally noted that, after 3 years, patients who initially experienced delayed graft function and received TAC/MMF survived at a higher rate (84.1%) than those who received CYA/MMF (49.9%, P = 0.02). This finding suggests that higher risk patients may derive benefit from receiving TAC over CYA. In the present study, for TAC efficacy analysis, the only appropriate groups to compare are the CYA/MMF and the TAC/MMF groups, since MMF has been shown to be more effective than AZA at preventing rejection. This study, which used modified CYA, did not find a difference in acute rejection rates as had the previously discussed European and US studies. The numbers of patients compared in the TAC/MMF and CYA/MMF groups were much smaller than those studied in the US and European randomised controlled trials. This may have resulted in a lack of power to detect a difference in acute rejection rates between modified CYA- and TAC-treated groups.
 Taking all of these findings together, it appears that TAC is at least as effective as modified CYA and may be more effective at preventing acute rejection.
Taking all of these findings together, it appears that TAC is at least as effective as modified CYA and may be more effective at preventing acute rejection.Mycophenolate mofetil (MMF)
MMF was approved in 1995 for the prevention of acute rejection in kidney transplant patients. MMF is a prodrug which is hydrolysed immediately in the bloodstream to its active form of mycophenolic acid (MPA). In 2004, an enteric-coated dosage form of mycophenolate sodium was also approved. MPA is the active moiety of both preparations. MPA prevents lymphocyte activation through its inhibition of inosine monophosphate dehydrogenase, an important enzyme in the production of guanosine nucleotides for lymphocyte DNA formation.22 Since B and T lymphocytes require de novo synthesis of purines, such as guanosine, MPA has potent cytostatic effects on these cell lines. When administered orally, MMF exhibits approximately 94% bioavailability in healthy volunteers. In kidney transplant recipients, bioavailability improves as patients progress further from the time of transplant. By month 3 following kidney transplant, bioavailability is similar to that of a healthy volunteer.
MMF was originally approved based on several randomised double-blind trials, all of which showed decreased treatment failure (biopsy-confirmed acute rejection, graft loss, patient death or discontinuation of study drug) at 6 months post-kidney transplantation with MMF when compared with either placebo or AZA.23–25 The US MMF Study Group and the Tricontinental MMF Study Group both enrolled approximately 500 cadaveric renal transplant recipients and randomised them to receive either MMF 2 g/day, MMF 3 g/day or AZA 1–2 mg/kg/day. All patients also received CYA and corticosteroids as their maintenance immunosuppression. The primary end-point of each study was BPAR or treatment failure. The initial reports from these studies analysed outcomes at 6 months.
 In each case, both MMF groups were shown to be superior to AZA, with substantially fewer patients in the MMF groups reaching the primary end-point when compared to AZA.
In each case, both MMF groups were shown to be superior to AZA, with substantially fewer patients in the MMF groups reaching the primary end-point when compared to AZA.The European MMF Cooperative Group, US MMF Study Group and Tricontinental Study Group all reported 3-year follow-up results with particular interest in patient and graft survival.26–28 Neither of the active control trials (US trial or the Tricontinental trial) reported differences in patient or graft survival at 3 years. However, the European trial did report improved 3-year graft survival (excluding death with a functioning graft) in the MMF 2 g group compared with the placebo group. The graft loss rate in the placebo group was 16% compared with 8.6% in the MMF 2 g group (P = 0.03).
With the exception of the Tricontinental placebo-controlled trial MMF 2 g group, in each of the above randomised trials, although MMF was shown to produce superior outcomes with fewer patients experiencing treatment failure when compared with either AZA or placebo, these studies showed no improvement in graft survival. This is likely due to lack of power in the above studies to detect such a difference. In an effort to determine whether or not MMF use was related to improved graft survival, Ojo et al. examined the United States Scientific Transplant Registry and compared patients treated with MMF with those treated with AZA between 1992 and 1997.29 The primary end-point is this study was late (more than 6 months post-transplantation) renal allograft failure. A multivariate analysis including potential risk factors for chronic allograft failure was performed on 66 774 patients who received a CNI, corticosteroids and either AZA or MMF; 48 436 patients received azathioprine, 8435 received MMF and 9903 received a mixture of different maintenance regimens. Patients were analysed based on their medication assignment at the time of discharge from transplant surgery. Although not a primary aim of the study, the analysis detected a higher rate of acute rejection in AZA-treated patients (24.7%) compared with MMF-treated patients (15.5%, P < 0.001). Graft survival (death censored) at 4 years was higher among the MMF group (85.6%) compared with the AZA group 81.9% (P < 0.0001) (Fig. 5.2). Patient survival at 4 years was also higher in the MMF group (91.4%) compared with 89.9% in the AZA group (P = 0.002). The risk factor most strongly associated with late graft loss was the presence of acute rejection, with a risk ratio of 2.41 (P < 0.001).
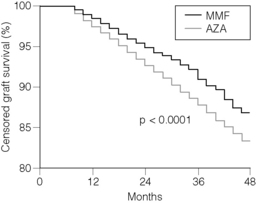
Figure 5.2 Comparison of Kaplan-Meier 4-year censored graft survival beyond 6 months after transplantation.
Reproduced from Ojo AO, Meier-Kriesche HU, Hanson JA et al. Mycophenolate mofetil reduces late renal allograft loss independent of acute rejection. Transplantation 2000; 69:2405–9. With permission from Lippincott, Williams & Wilkins.

Sirolimus
Sirolimus, the first of a new class of immunosuppressive medications, was approved in 1999. It belongs to the therapeutic class, mammalian target of rapamycin (mTOR) inhibitors. Like other immunosuppressive agents, sirolimus inhibits T-lymphocyte activation and proliferation, but does so through a unique mechanism. Sirolimus, like TAC, binds to the intracellular FKBP12, but the sirolimus–FKBP12 complex results in different downstream effects than the TAC–FKBP12 complex. The sirolimus–FKBP12 complex binds and inhibits mTOR. mTOR is a key enzyme, allowing the T lymphocyte to respond to cytokine-initiated cell proliferation and move from the G1 to the S phase of the cell cycle.30
Sirolimus exhibits important pharmacokinetic differences from other commonly used immunosuppressive medications, and also undergoes clinically important drug–drug interactions. Sirolimus is administered once daily and exhibits bioavailability of approximately 14% as the oral liquid formulation and approximately 40% as oral tablets. Despite this difference in bioavailability it is recommended that 2 mg of sirolimus oral solution and 2 mg sirolimus administered as tablets are clinically equivalent and may be interchanged 1:1 when formulation change is necessary.30 This has not been confirmed for higher doses of sirolimus. When administered with CYA, it is recommended that sirolimus be separated from CYA administration by 4 hours to avoid an approximately 45% increase in sirolimus exposure.31 This interaction has not been shown when used in combination with TAC. Although serum concentration monitoring was not initially thought to be important with sirolimus, this practice is now routine. This therapeutic drug monitoring is accomplished through 24-hour trough concentration monitoring. Like CYA and TAC, the therapeutic range of sirolimus trough concentrations depends largely on other immunosuppressive medications and patient-specific factors, but in general the therapeutic range is 5–15 ng/mL. Sirolimus is primarily metabolised by cytochrome P450 3A4. Concomitant administration with medications that inhibit or induce this microenzyme should result in dosage adjustments and increased trough concentration monitoring of sirolimus.
Sirolimus was approved for the prevention of acute rejection in kidney transplant recipients based on the results of two randomised, double-blind trials.32,33 Each trial randomised patients to one of three groups: a 2 mg/day sirolimus group, a 5 mg/day sirolimus group or an AZA group (US trial) or a placebo group (worldwide trial). Each trial also used CYA and prednisone as additional maintenance immunosuppression. Neither study allowed antibody induction treatment at the time of transplantation. The primary composite end-point of each study was the rate of efficacy failure (BPAR, graft loss or death) at 6 months post-transplantation.
 This finding of apparent synergistic nephrotoxicity with full-dose CNI and sirolimus has been replicated in another randomised controlled trial comparing TAC/sirolimus vs. TAC/MMF.34
This finding of apparent synergistic nephrotoxicity with full-dose CNI and sirolimus has been replicated in another randomised controlled trial comparing TAC/sirolimus vs. TAC/MMF.34
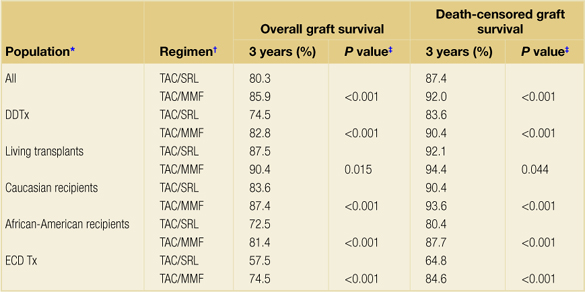
In addition, analyses of the United States Scientific Renal Transplant Registry showed lower 4-year death-censored graft survival with CYA/sirolimus immunosuppression as compared with CYA/MMF immunosuppression, and 3-year death-censored graft survival with a TAC/sirolimus immunosuppression as compared with TAC/MMF in deceased donor transplants35,36 (Table 5.1). These outcomes lead to the practice of no longer using sirolimus with full-dose CNI, but rather exploring the use of sirolimus in various CNI-sparing regimens. These regimens will be discussed later in this chapter.
Table 5.1Overall and death-censored graft survival in tacrolimus/mycophenolate mofetil and tacrolimus/sirolimus regimens
 Also, since the initial pivotal trial publication, problems with impaired wound healing and increased incidence of lymphoceles have been noted with sirolimus.
Also, since the initial pivotal trial publication, problems with impaired wound healing and increased incidence of lymphoceles have been noted with sirolimus.Contribution of antibody products for induction immunosuppression and for treatment of acute rejection episodes
Antibody therapies are generally used for two indications following kidney transplantation: induction treatment and treatment of acute rejection episodes. Induction immunosuppressive treatment occurs at the time of transplantation and refers to the use of an antibody preparation to provide increased immunosuppression during and immediately after transplantation. Antibody therapy for the treatment of acute rejection is generally selected after high-dose corticosteroid failure.
Depleting antibody preparations
Depleting antibody preparations may be used both for the treatment and the prevention (as induction immunosuppression given at the time of transplant) of acute rejection in kidney transplantation. There are four primary depleting antibody preparations in use to date. These agents can be further subdivided into monoclonal and polyclonal agents. The two available polyclonal products are equine antithymocyte globulin (E-ATG) and rabbit antithymocyte globulin (R-ATG). The two monoclonal products are muromonab CD3 and alemtuzumab. Muromonab CD3 is a chimeric monoclonal antibody, whereas alemtuzumab is a humanised monoclonal antibody. These four depleting antibody agents will be discussed in the order in which they became available for use, with E-ATG approved in 1981, muromonab CD3 in 1986, R-ATG in 1999 and alemtuzumab (used off-label in organ transplantation) approved for treatment of chronic lymphocytic leukaemia in 2001.
Equine antithymocyte globulin
E-ATG was the first depleting or non-depleting antibody to receive approval in kidney transplantation. It is currently approved for the management of allograft rejection in renal transplant patients.37 E-ATG is a polyclonal IgG product produced by immunising horses with human T lymphocytes. E-ATG is thought to exert its antirejection effects by clearing T lymphocytes from peripheral blood, and modulation of T-lymphocyte activity. When used for treatment of acute rejection, E-ATG is usually administered in doses of 15 mg/kg intravenously daily for 14 days. E-ATG should be administered into a high-flow central vein through a 0.2–1 μm in-line filter. The manufacturer of E-ATG recommends administration of an intradermal test dose of 0.1 mL of a 1:1000 dilution (5 μg horse IgG). Although still recommended by the manufacturer, this practice has not been shown to accurately predict which patients may experience allergic reactions to E-ATG.
E-ATG was originally studied in 358 renal transplant recipients in an open-label study design as induction immunosuppression.38 Patients were randomly assigned to receive either E-ATG (n = 183) for 14 days at the time of transplant or no E-ATG (n = 175) along with their maintenance immunosuppression consisting of AZA and prednisone. E-ATG doses were between 10 and 30 mg/kg/day. The outcomes of interest were: time to first rejection episode, steroid dosage requirements, functional graft survival and patient survival. When all four study lots of E-ATG were combined for data analysis, there was no difference between patients who received E-ATG and those who did not in any parameter except time to first rejection episode. For approximately the first 28 days after transplant, the E-ATG group experienced fewer rejection episodes than did the control group. After this period of time the occurrence of rejection became similar between the groups. Thus, active dosing of E-ATG conferred protection from acute rejection, but not long after dosing ended, rejection rates in the treatment group resembled those of the control group.
Several months later, Shield et al. published their experience using E-ATG for treatment rather than prevention of rejection.39 Twenty patients with established rejection were randomised to receive E-ATG 15 mg/kg (doses were then adjusted based on presence of T-cell function measured by sheep red blood cell rosetting levels) for 14 days or methylprednisolone 1000 mg i.v. daily for up to 5 days. As in the previous study, maintenance immunosuppression consisted of AZA and prednisone in both groups. Both treatments successfully reversed rejection episodes in these 20 patients, with E-ATG reversing rejection on average in 3 days and methylprednisolone in 6 days. This early experience, although underpowered to detect any difference in efficacy between the two agents, seemed to prove that E-ATG was at least as effective as high-dose methylprednisolone and might reverse rejection episodes more quickly.
Given that E-ATG appeared at least as effective at treating rejection as high-dose corticosteroids, Hardy et al. investigated in a small group of patients the effect of E-ATG on allograft rejection that was resistant to standard high-dose corticosteroids.40 The authors’ typical treatment for acute rejection was methylprednisolone 15 mg/kg/day (not to exceed 750 mg) plus graft irradiation on days 1, 3 and 5 after rejection diagnosis. Ten patients who experienced acute rejection but did not respond to this typical treatment were treated with E-ATG 15 mg/kg/day for 21 days. Their response to this treatment was then described and compared to that of 10 other patients who were treated with and responded to the typical rejection protocol. The authors were particularly interested in whether or not the experimental group showed a return of SCr to baseline, the frequency of other rejection episodes following E-ATG treatment, 1-year graft survival and patient mortality. Although the patients in the experimental group did not respond initially to therapy as did the control group, seven of these patients’ SCr returned to pre-rejection concentrations compared to only three patients in the control group. Only two patients in the experimental group experienced a second rejection episode within 1 year and no patient in the experimental group experienced a third rejection episode. In contrast, four patients in the control group experienced a second rejection episode and two of these patients experienced rejection a third time within 1 year. SCr concentrations also appeared to be lower in the experimental group compared with the control group 1 year later. Five patients in the experimental group had SCr less than or equal to 1.5 mg/dL compared with two patients in the control group. Graft failure appeared to occur equally among the two groups, with five graft failures in the control group and four in the experimental group over the 1-year follow-up. No patient in either group died during the year after rejection diagnosis. Later, this same group of investigators published their experience with 12 additional patients receiving E-ATG for their first acute rejection and 20 control patients receiving the standard therapy for their first acute rejection.41 In this series graft survival at 1 year was 73% in patients who received E-ATG for rejection treatment compared with 46.6% in patients who received standard therapy. Mortality was 3% in the E-ATG group compared with 20% in the standard therapy group. This follow-up study appeared to suggest that treatment of acute rejection episodes with E-ATG rather than high-dose corticosteroids may confer better graft survival when used on the background of AZA/corticosteroid maintenance immunosuppression. Not long after E-ATG was approved for use, however, CYA was also approved as maintenance immunosuppressive therapy. As such, these same outcomes with E-ATG may not exist when stronger maintenance immunosuppressive therapy, including a CNI, is used.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


