CHAPTER 21 Vulvar Ulcers
Ulcers are differentiated from erosions on the basis of depth. Erosions are shallow and are due to absence of the epidermis with no appreciable damage to the underlying dermis. For this reason erosions have a red base or are covered with loosely adherent yellow crust. Ulcers are deep with the defect extending into the dermis. The base of an ulcer contains necrotic tissue, blood-tinged crust, or blue-black adherent eschar. Only vulvar ulcers are discussed in this chapter; erosive vulvar disease is covered in Chapter 20.
Aphthous ulcer and complex aphthosis
Epidemiology and clinical manifestations
Gynecologists and dermatologists generally believe that aphthous ulcers occur only rarely on the vulva. The medical literature seems to support this belief. Even though the first patients with noninfectious vulvar ulcers of this type were reported by Lipschutz as far back as 1913, there are only a handful of such patients described in published reports. Only a single manuscript describes incidence or prevalence rates1. However, our own experience, along with discussion among colleagues, suggests that aphthous ulceration of the vulva occurs with some frequency. The risk factors for development of vulvar ulcers of this type are the same as those described for oral disease and, similarly, recurrences are common.
The best data regarding vulvar aphthous ulcers are found in the recent report by Huppert et al.1 These authors described 20 females with idiopathic vulvar ulcers. The mean age of the patients was 14 years (range 10–19 years). Most ulcers were less than 1 cm in diameter but one patient had a 5-cm lesion. Multiple lesions were almost always present, occurring in 17 of 20 patients. The most common site was within the vestibule but ulcers were also occasionally found on the keratinizing epithelial surfaces of labia majora and perineum. (This is in marked distinction to oral aphthae, which are never found on the perioral skin of the face.) Half of their patients reported oral lesions and one-third developed recurrences of their vulvar ulcers. My experience is very similar, though a larger proportion of my patients have had oral lesions and multiple vulvar recurrences.
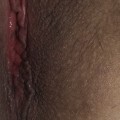
Individual lesions start out as round to oval ulcers but coalescence of neighboring lesions can result in larger ulcers with serpiginous borders (Figures 21.1–21.5). A red rim of erythema (2–3 mmwide) is present and, at the base of the ulcers, there is usually a white coagulum of necrotic tissue. The ulcers are quite painful and healing generally requires 2–3 weeks. Scarring can occur but it seems less severe than would be expected given the size and depth of the lesions.
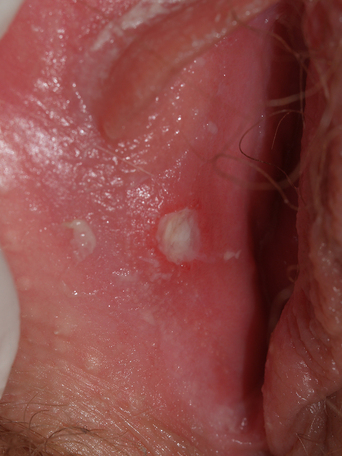
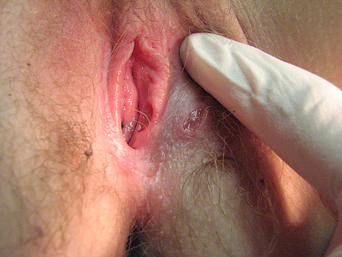
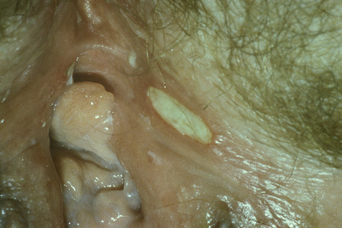
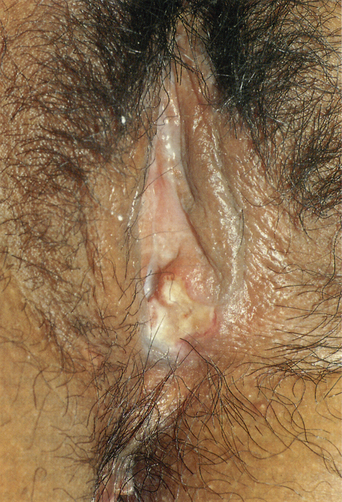
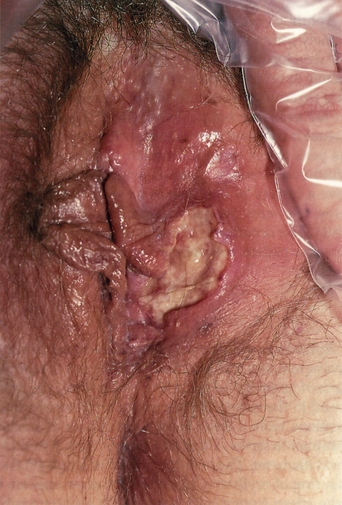
Figure 21.5 Very large aphthae sometimes heal with considerable scarring.
(Reproduced from P. J. Lynch and L. Edwards Genital Dermatology (Fig. 18-2), New York: Churchill Livingstone; 1994.)
Systemic symptoms and signs such as fever, malaise, headache, gastrointestinal distress, pharyngitis, and myalgia occur in many of these young women with complex aphthosis but these are usually mild, time-limited, and nondebilitating1.
Diagnosis and differential diagnosis
The appearance of these aphthous ulcers is sufficiently distinctive that, in association with a history of recurrence, a clinical diagnosis is possible. Since there are no microscopic or laboratory features that positively identify the disease, the only way to confirm a clinical diagnosis is to rule out conditions that simulate it (Table 21.1). Genital herpes (herpes simplex virus: HSV) is almost always a consideration but, with the exception of chronic HSV infection in the immunosuppressed, the lesions of herpes are not as large or deep. However, in some instances, immunofluorescent smears, culture, and biopsy may be necessary to rule out herpetic infection. Primary syphilis (chancre) can be identified by serology and biopsy. Cultures and/or biopsy are appropriate to rule out the anogenital ulcers of Crohn’s disease as well as the lesions of chancroid, granuloma inguinale, and various cutaneous malignancies.
Table 21.1 Aphthous Ulcers and Complex Aphthosis
| Diagnosis |
| Painful, deep ulcers |
| No undermining of edges |
| Thin red border |
| White coagulum at the ulcer base |
| History of recurrence |
| Differential Diagnosis |
| Herpes simplex virus in the immunosuppressed |
| Crohn’s disease |
| Primary syphilis |
| Therapy |
| Rule out associated disease, especially Behçet’s disease |
| Lidocaine or silver nitrate for pain |
| Topical or intralesional steroids |
| Oral antibiotics as anti-inflammatory agents |
| Consider short-term systemic steroids |
| Consider dapsone, colchicine, thalidomide |
As indicated above, complex aphthosis is the term used when vulvar aphthous ulcers occur in association with either a synchronous or metachronous history of oral aphthous ulcers. Patients with complex aphthosis and no related underlying disease are said to have primary complex aphthosis. When patients do have an associated systemic disorder, the term “secondary complex aphthosis” should be used.
Histology and laboratory tests
Because there are several medical problems that can be associated with complex aphthosis (see above), a complete medical history must be taken. Laboratory tests are then chosen based on a history of specific problems. Letsinger et al. have recently suggested specific steps for laboratory evaluation2.
Pathogenesis
The cause of aphthous ulceration is not known. Based on the high frequency of a positive family history, genetic factors are probably important. It has been hypothesized that inheritance involves the predisposition to develop a more brisk than normal cell-mediated immune response to one or another unrecognized antigen. Although the nature of the antigen(s) has not been identified, there is some evidence to suggest that microbial proteins, by way of molecular mimicry, initiate an autoreactive (autoimmune) response. In any event, activation of the cell-mediated immune response results in the generation of many inflammatory cytokines, such as tumor necrosis factor-alpha and interleukin-2. Both the activated immune cells and the cytokines they release are presumably cytotoxic to epithelial cells and possibly also to fibroblasts. Risk factors for the development of aphthous ulceration are discussed above. Specific triggers to the development of individual attacks include local tissue trauma (pathergy), psychological stress, and possibly acidic and salty foods.
Therapy and prognosis
Letsinger et al. have developed a widely accepted therapeutic ladder that starts with topical agents (local anesthetics, local steroids, and tacrolimus) and progresses to systemic therapy with oral colchicine, dapsone, a combination of these two agents and, if necessary, thalidomide2. However, as indicated in their manuscript, only about 5% of patients respond satisfactorily to topical agents alone. On the other hand, with systemic therapy, 88% of their patients achieved 50% or greater improvement2.
I have found that topical anesthetics such as 5% lidocaine ointment and EMLA are of limited use. For the reduction of pain in small lesions I prefer the application of silver nitrate using standard silver nitrate sticks3. Clobetasol 0.05% ointment and gel are only modestly effective anti-inflammatory agents. Intralesional injection with triamcinolone in a concentration of 10 mg/mL works much better but such therapy is painful and requires an office visit for each recurrence.
My initial treatment of choice is a brief “burst” of oral prednisone in a dose of 40 mg q a.m. for 7 days. Oral antibiotics, used for their anti-inflammatory properties, such as doxycycline 100 mg bid or minocycline 100 mg bid, can be started at the same time and may be continued indefinitely in an attempt to prevent the recurrence of additional lesions. Colchicine, 0.06 mg bid to tid, dapsone 100–150 mg/day, and pentoxyphylline 400 mg tid are useful alternatives to antibiotics4,5. Thalidomide and other tumor necrosis factor-alpha inhibitors are remarkably effective6,7 but adverse side-effects, high cost, and relatively short history of their use limit this approach to patients who have failed more conventional therapy.
The prognosis for women with idiopathic (primary) complex aphthosis involving the vulva is unclear. The medical literature on Behçet’s disease states that patients may have oral and genital lesions (complex aphthosis) for several years prior to the development of systemic symptoms and signs. In the series reported by Letsinger et al., 15% of patients with complex aphthosis referred to their group met established criteria for the diagnosis of Behçet’s disease2. However, no information was provided in that paper as to whether the complex aphthosis preceded the Behçet’s disease or, if so, by how long. Importantly, none of the 20 patients studied by Huppert et al. had or developed Behçet’s disease when followed over a rather short mean period of 14 months1. Finally, based on my own experience and on discussion with other vulvologists, I believe that progression to Behçet’s disease is extremely unlikely in women of European or North American background. Thus, when the standard diagnostic criteria for Behçet’s disease are not met, I feel that it is inappropriate to suggest that patients have, or are likely to develop, Behçet’s disease (see also discussion in the section on Behçet’s disease, below).
Behçet’s disease
Women with complex aphthosis (see above) already have recurring oral and vulvar ulcers and thus need only one other criterion before they meet the ISG diagnostic guidelines for Behçet’s disease. Unfortunately, in western societies, the skin lesions listed in the ISG criteria are quite common as “stand-alone” conditions and they also occur with appreciable frequency in patients with inflammatory bowel disease, lupus erythematosus, and other autoimmune disease. For this reason, many women in the United States, continental Europe, and the United Kingdom meet the ISG criteria for Behçet’s, but do not develop the severe ocular or central nervous system disease that characterizes eastern patients with Behçet’s disease. For this reason, one must be extremely cautious about telling a woman with complex aphthosis and mild skin or eye findings that she has, or is likely to develop, Behçet’s disease.
Crohn’s disease
Inflammatory bowel disease (Crohn’s disease and ulcerative colitis) is quite common in North America and Europe, with a prevalence of around 400 per 100 000 persons. Crohn’s disease occurs somewhat less frequently than ulcerative colitis. It has an annual incidence rate of about 8 per 100 000 and a prevalence of about 150–175 per 100 0008. However, cutaneous lesions are found more often in Crohn’s disease, occurring in about 2–3% of such patients9. Patients with ulcerative colitis can develop perianal fistulae but other types of anogenital involvement are rare, if they occur at all. For the purposes of this section, discussion will be restricted to Crohn’s disease.
Epidemiology and clinical manifestations
Although histologically specific mucocutaneous lesions of Crohn’s disease can occur anywhere on the body, most occur in the anogenital area. Ploysangam et al., in the largest available review, identified 80 children who had cutaneous Crohn’s disease10

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


