dsDNA viruses
Poxviridae
Molluscum contagiosum virus
Herpesviridae
α: human herpesvirus 1, 2, 3 (HSV-1, HSV-2, VZV)
β: human herpesvirus 5, 6, 7 (HCMV, HHV-6, HHV-7)
γ: human herpesvirus 4, 8 (EBV, KSHV)
Papillomaviridae
Human papillomavirus (HPV) 1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 57, 60
Smallpox
Vaccinia virus (VV)
ssDNA viruses
Parvoviridae
Human parvovirus B19
DNA and RNA reverse transcribing virus
Retroviridae
Human T-lymphotropic virus 1 (HTLV-1)
Lentivirus
Human immunodeficiency virus 1 (HIV-1)
Negative stranded ssRNA viruses
Rhabdoviridae
Rabies virus
Paramyxoviridae
Measles virus
Positive stranded ssRNA viruses
Picornaviridae
Human enterovirus A, Coxsackie virus A
Flaviviridae
Dengue virus (DV), Japanese encephalitis virus (JEV), West Nile virus (WNV)
Togaviridae
Rubella virus
This chapter summarizes the current knowledge of the host antiviral immune systems in the skin and the recent advances regarding cutaneous antiviral immunity, highlighting the innate immunity against the viruses that are transmitted via skin or associated with cutaneous symptoms.
19.2 Antiviral Innate Immunity
19.2.1 Viral Recognition by Skin Cells
In the skin, the PAMPs of invading pathogens are recognized by host innate receptors known as PRRs located both at the skin cell surface and within the skin cells (see also Chaps. 17, 18, and 19). Viral recognition by the innate immune system is more challenging than recognition of other pathogen classes, because any given viral protein is unlikely to be shared among diverse viruses [1, 2]. However, remarkable progress has been made over the past few years towards understanding the contribution of PRRs, such as C-type lectins (CLRs), Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I like receptors (RLRs), and other cytosolic PRRs, to viral detection.
In general, the detection of viral PAMPs via PRRs initiates two major innate immune signaling cascades: the first involves the activation of the transcription factors interferon regulatory factor 3 (IRF3) and/or IRF7, nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1), which cooperate to induce the transcription of type I interferons (IFNs, e.g., IFN-α and IFN-β), chemokines and pro-inflammatory cytokines (Figs. 19.1 and 19.3). Through the secretion of type I IFNs, the response can be amplified and spread to surrounding uninfected skin cells and thereby activate hundreds of interferon-stimulated genes (ISGs) , most of which encode products with profound antiviral effects, such as the degradation of viral nucleic acids or inhibition of viral gene expression [3, 4]. The second signaling pathway results in the formation of an inflammasome complex, which activates caspase-1, a protease which processes pro-interleukin (IL) 1-β and pro-IL–18 to generate active cytokines ready for secretion (Fig. 19.2). In general, TLRs and RLRs are involved in the expression of type I IFNs or pro-inflammatory cytokines and chemokines, whereas viral detection by NLRs leads to caspase-1–mediated processing of IL-1β. The importance of viral recognition via these PRRs and subsequent cytokine production is illustrated by the extreme susceptibility to percutaneous viral infection of mice lacking the PRRs and the cytokines.
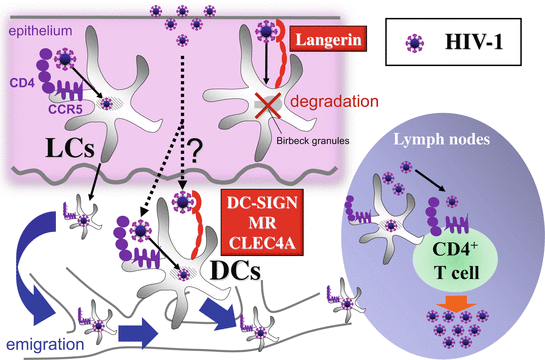
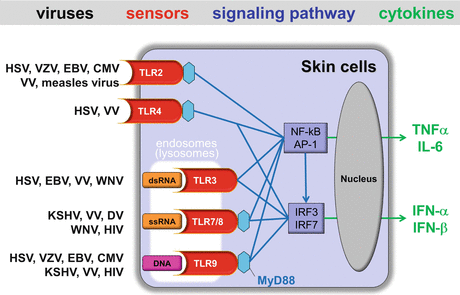
Fig. 19.1
CLRs-mediated HIV-1 recognition and initial biologic events in sexual transmission of HIV. Langerhans cells (LCs) within epithelium of genital mucosa or skin are infected with invading HIV-1 via CD4 and CCR5, and then emigrated to the draining lymph nodes, where they replicate the virions and transmit the virus to CD4+ T cells, leading to vigorous production of new HIV virions by T cells (cis-infection). HIV-1 bound to langerin is internalized within birbeck granules, where the virus is degraded. DC-SIGN, MR, and CLEC4A promote cis-infection of DCs and also facilitate trans-infection to CD4+ T cells
Fig. 19.2
TLRs-mediated viral recognition and signaling pathways in skin cells. Upon ligand stimulation by the indicated skin-associated viruses, TLRs, except the TLR3, recruit the adaptor MyD88. This allows NF-κB to translocate into the nucleus and simultaneously activates the MAP kinase pathway triggering the activation of AP-1. Together, NF-κB and AP-1 induce the expression of pro-inflammatory cytokines. Upon viral ligand stimulation, TLR7 and TLR9 associate with MyD88 leading to nuclear translocation of IRF7. Stimulation of TLR3 and TLR4 permits nuclear translocation of IRF3. IRF3 or IRF7, along with NF-kB and AP-1, cooperate to induce the expression of type I IFNs, IFN-β, and IFN-α, respectively
19.2.2 C-Type Lectin Receptors (CLRs)
CLRs play a pivotal role in the immune system as pattern-recognition and antigen-uptake receptors. CLRs are an important family of PRRs and involved in the recognition of certain skin-associated viruses, which express high-mannose structures (see Chap. 18). The cutaneous innate immune cells can bind skin-associated viruses via CLRs, such as DC-specific ICAM-3–grabbing nonintegrin (DC-SIGN ; CD209) [5–7], langerin (CD207) [8, 9], Mannose receptor (MR; CD206) [10, 11], DEC-205 (CD205) [12], C-type lectin-like receptor (CLEC) 4A (also known as DC immunoreceptor; DCIR) [13, 14], CLEC 5A [15, 16], and CLEC9A (also known as DNGR-1) [17, 18] (Table 19.2). These CLRs recognize mannose, fucose, and glycosylated structures of the viruses via conserved carbohydrate recognition domains. CLRs have endocytic activity, mediate internalization of the viral ligand or the virus itself, and can direct cargo into intracellular compartments that allow for antigen processing/presentation and the induction of specific gene expression profiles to the viruses, either by modulating TLR signaling or by directly inducing gene expression (see Chap. 18). Although most CLRs do not act as “self-sufficient” PRRs for the viruses, the few CLRs trigger distinct signaling pathways via a number of kinases, including spleen tyrosin kinase (SYK) and Src kinase, that modulate the induction of specific cytokines [19, 20]. In addition, recent studies have suggested that several CLRs, such as DC-SIGN and CLEC4A, induce signaling pathways that modulate TLR-induced gene expression at the transcriptional or posttranscriptional level [19, 21]. For example, the interaction of DC-SIGN with mannose-containing virus, such as human immunodeficiency virus (HIV-1) and measles virus, affects TLR4-mediated immune responses by DCs [22]. The crosstalk between TLR4 and DC-SIGN depends on the prior activation of NF-κB by TLR signaling and is therefore not limited to TLR4, but also includes triggering of other NF-κB–inducing PRRs, such as TLR3 and TLR5 [22].
Table 19.2
C-type lectin receptors and related skin-associated viruses
CLRs | Viral Ligands | Expression |
|---|---|---|
DC-SIGN (CD209) | HIV-1, CMV, DV, measles virus | Myeloid DCs, macrophages |
Langerin (CD207) | HIV-1, measles virus | Langerhans cells (dermal DCs) |
Mannose receptor (CD206) | HIV-1, DV | Myeloid DCs, macrophages |
DEC-205 (CD205) | HIV-1 | Myeloid DCs |
CLEC4A (DCIR) | HIV-1 | Myeloid DCs, macrophages, pDCs |
CLEC5A | DV, JEV | Monocytes, macrophages |
CLEC9A (DNGR-1) | HIV-1 | Myeloid DCs |
CLRs not only act as beneficial to the host, but also contribute viral spread and pathogenicity. Most studies for CLRs in cutaneous viral infection have long been focused on their roles in the sexual transmission of HIV-1. HIV-1 can bind various CLRs expressed by cutaneous innate immune cells (Table 19.2), which also express CD4 and coreceptors (CCR5 and CXCR4), required for HIV-1infection (Figs. 19.1 and 19.4) [19, 20]. There are two major mechanisms of HIV-1 transmission between cells; one is cis-infection, where target cells are infected with progeny virions, which are released by productively infected cells, via CD4 and coreceptors, and the other is trans-infection, where target cells are infected with virions, which were captured by the neighboring donor cells without productive infection, via the CLRs-mediated virological synapse or the exosome-like pathway [19]. Of note, cis and trans pathways via CLRs are not mutually exclusive, because, in the cis-infection, CLRs can facilitate de novo infection of donor cells. Indeed, DC-SIGN, MR, and CLEC4A promote cis-infection of innate immune cells through increased interactions between HIV-1 glycoprotein envelope gp120 and CD4, but also facilitate viral capture and CD4+ T-lymphocyte trans-infection by mediating viral endocytosis into nondegradative endosomes permitting the intracellular storage of intact virions [19, 20]. DC-SIGN–mediated signaling is even more complex, as HIV-1 requires signaling by DC-SIGN for the replication and the generation of full-length viral transcripts in DCs [23]. In contrast, Langerin impairs infection of LCs by HIV-1 via subsequent internalization within birbeck granules, where the virus is degraded (Fig. 19.1) [8]. Nevertheless, there is a saturation of langerin at higher virus concentrations that overwhelms the protective mechanism of action so that langerin ultimately is unable to completely prevent LC infection [8] and CD4/CCR5-mediated cis-infection of LCs is considered to be a major pathway involved in the sexual transmission of HIV-1 (Fig. 19.1) [24–26]. In fact, LCs can be productively infected by HIV-1 ex vivo and transmit the virus to cocultured CD4+ T cells [27–29]. Importantly, consistent with the preferential sexual transmission of R5 HIV-1 (CCR5-utilizing HIV), LCs are preferentially infected by R5 HIV, but not X4 HIV (CXCR4-utilizing HIV) [27, 28, 30]. Moreover, it has been observed that up to 90% of initially infected target cells were LCs in Rhesus macaques exposed to intravaginal simian immunodeficiency virus (SIV), and topical application of CCR5 inhibitors completely protected Rhesus macaques from intravaginal exposure of SHIV, a chimeric simian/human immunodeficiency virus, whereas the CLR-inhibitor, mannan, could not [31–33].
19.2.3 Toll-Like Receptors (TLRs)
Several endosomal members of the TLRs family such as TLRs-3, -7, -8, and -9 can recognize nucleic acids of both DNA and RNA viruses (Table 19.1). Viral double-stranded (ds) RNA (dsRNA), single-stranded (ss) RNA (ssRNA), and unmethylated CpG-rich motif in DNA can be recognized by TLR3, TLR7/TLR8, and TLR9, respectively, and all of these nucleic acid-sensing TLRs induce the nuclear translocation of IRF3 and/or IRF7, leading to the expression of type I IFNs (Fig. 19.2) (see also Chap. 17) [1]. The viral ligand stimulation of TLR7/TLR8 and TLR9 recruits the adaptor MyD88, allows NF-κB to translocate into the nucleus, and simultaneously activates the MAP kinase pathway triggering the activation of AP-1. Together, NF-κB and AP-1 induce the expression of pro-inflammatory cytokines. Because viral particles are generally endocytosed and degraded in late endosomes or lysosomes, viral DNA and RNA are released into these intracellular acidic compartments, allowing viral nucleic acids to be in close contact with endosomal TLRs. Alternatively, nucleic acids present in the cytoplasm may be engulfed by an autophagosome, which subsequently fuse with the endosome, and are recognized by the TLRs [34].
dsRNA that is produced by most viruses during their replicative cycle is often considered as a PAMP [35]. RNA viruses are important producers of dsRNA, however, only a few RNA viruses have been shown to induce a TLR3 -dependent innate immune response [36]. During West Nile virus (WNV), a mosquito-transmitted flavivirus infection, TLR3-mediated inflammatory responses by viral dsRNA contribute to the development of lethal encephalitis by facilitating virus entry into the brain [37]. TLR3 is hypothesized to enable recognition of a DNA virus because virtually all viruses produce dsRNA at some point during replication [38]. Indeed, in several skin-associated DNA virus infections, TLR3 has been shown to play a role in the antiviral response (Fig. 19.2). For example, it has been demonstrated that herpes simplex virus types 1 (HSV-1) activates keratinocytes to produce type I IFN in a TLR3-dependent manner [39] and TLR3 is required to control HSV-1 spreading to the central nervous system [40]. Activation of TLR3 by viral dsRNA induces not only protective effects against viral infection but also effects that contribute to viral pathogenesis. In Epstein–Barr virus (EBV)-associated hemophagocytic lymphohistiocytosis and infectious mononucleosis, TLR3 contributes to the sudden release of inflammatory cytokines, as the release of EBV-encoded small RNA (EBAR) from EBV-infected cells, which are giving rise to dsRNA-like molecules, activate immune cells via TLR3 signaling to produce type I IFNs and TNF-α [41]. New insights have recently emerged regarding the TLR3-mediated recognition of viral dsRNA; Class A scavenger receptors bind to extracellular viral dsRNA, which is released into the extracellular space after lysis of infected cells and mediate uptake and presentation of dsRNA to TLR3 in the endosome [42]. As well as TLR3, a recent study revealed that TLR4 stimulation also promotes activation of IRF3 and signal to NFκB [43]. Indeed, TLR4 has been shown to be important for the induction of antiviral cytokines in the response to HSV and vaccinia virus (VV) (Fig. 19.2) [2].
TLR7 and TLR9 detect viral RNA and DNA, respectively, in the endosomal lumen of virus-infected cells and TLR7- and TLR9-dependent signaling pathways require MyD88 and IRF7 to induce type I IFNs, especially IFN-α (see Chap. 17). TLR7/8 can be stimulated by several skin-associated viruses, including Kaposi’s sarcoma-associated herpesvirus (KSHV), VV, Dengue virus (DV), West Nile virus (WNV), and HIV-1, and TLR9 can sense all herpes viruses, VV, and HIV-1 (Fig. 19.2) [44]. Most skin cells are able to produce type I IFNs in response to viruses. However, it is of note that for these endosomal TLRs, induction of type I IFNs generally occurs in plasmacytoid DCs (pDCs), “professional IFN producers,” [44, 45] which are essentially absent in normal skin but have been described in lesions of inflammatory diseases of the skin (e.g., psoriasis vulgaris, contact dermatitis, and lupus erythematosus) [46]. Intriguingly, in mice models, after i.v. inoculation of HSV, TLR9-dependent recognition of CpG motifs in HSV DNA mediates IFN-α secretion by pDCs, whereas pDCs have a negligible impact on local type I IFN production after vaginal or cutaneous HSV infection [47]. Nevertheless, it is still possible that pDCs may play some roles in human anti-HSV immune responses, because pDCs have been shown to infiltrate the reactivated lesions of genital herpes abundantly and persist for months after healing [48]. HSV recognition by TLR9 is considered to be mediated through an endocytic pathway, but does not require HSV viral replication [49], suggesting that the interaction between HSV glycoproteins and cell surface receptors may induce HSV internalization and subsequent release of viral DNA in the endosomal compartment, allowing the interaction with TLR9 [36]. TLR7 has been shown to be involved in recognition of WNV following intradermal infection, and TLR7 activation by ssRNA of WNV promotes migration of epidermal LCs [50, 51]. In addition, deficiency in TLR7 dramatically impairs recognition of ssRNA and immune cell homing to infected target cells during infection with WNV, resulting in increased viremia and susceptibility to lethal WNV infection [52]. TLR7 has also proved to be relevant to episodic reactivation of latent Kaposi’s sarcoma-associated herpesvirus (KSHV) [53] and HIV pathogenesis through type I IFN production by pDCs [54]. A recent study has demonstrated that HIV-1 evolves a strategy to turn TLR8 signaling in DCs to its own advantage [23]. TLR8 triggering by HIV-1 ssRNA activated NF-κB, which was required for transcription initiation of the integrated HIV-1 genome by RNA polymerase II (RNAPII), and transcription elongation by RNAPII resulted in full-length HIV-1 transcripts and productive DC infection. Indeed, inhibition of TLR8-induced signaling prevented productive HIV-1 infection of DCs and subsequent transmission of HIV-1 to T cells. Thus, HIV-1 subverts the innate signaling pathways by TLR8 for its replication in DCs and subsequent transmission to T cells. Additionally, VV DNA can induce large amounts of type I IFNs in cells after recognition by TLR8 [55].
Recent studies have revealed that TLR2 can be stimulated by several skin-associated viruses, including HSV-1 and HSV-2 [56, 57], varicella-zoster virus (VZV) [58], human cytomegalovirus (HCMV) [59, 60], mouse cytomegalovirus (MCMV) [61, 62], measles virus [63], and VV [62, 64] (Fig. 19.2). In most of these cases, the recognition by TLR2, in combination with TLR1 or TLR6, recruits the adaptor MyD88, resulting in translocation of NF-κB into the nucleus and triggering the activation of AP-1, which cooperate to induce the expression of pro-inflammatory cytokines, thus acting as beneficial to the host and mounting an effective innate or adaptive immune response to viruses. For example, VZV and measles virus have been demonstrated to activate human monocytes to produce pro-inflammatory cytokines such as IL-6 in a TLR2-dependent manner [58, 63]. In addition, TLR2 and CD14 expressed on permissive human fibroblast cell strains recognize viral envelope proteins of HCMV, leading to induction of inflammatory cytokines and innate immune activation [60]. Surprisingly, although it is well known that bacterial ligands for TLR2 are unable to drive type I IFN production (see Chap. 17), VV and MCMV have recently been demonstrated to be able to induce type I IFNs via TLR2 [62], thus acting as beneficial to the host. Note that this specialized response requires TLR2 internalization and is mediated by a particular cell type (Ly6C, hi, inflammatory monocytes), which has been recently found to recruit to skin in allergic inflammation [65]. On the contrary, viral interaction with TLR2 can be detrimental to its host. HSV-1 recognition by TLR2 induces the expression of inflammatory cytokines in the brain that cause lethal encephalitis [57]. TLR2 activation by measles virus not only induces pro-inflammatory cytokines but also upregulates surface expression of CD150, the receptor for measles virus, indicating that activation of TLR2-dependent signals might contribute viral spread and pathogenicity [63].
19.2.4 NOD-Like Receptors (NLRs)
The cytoplasmic PRRs, nucleotide oligomerization domain (NOD)-like receptors (NLRs), have been recently implicated to recognize viral PAMPs. NLRs that contain a caspase activation and recruitment domain (CARD) are part of the NLRC subfamily including NOD2, whereas NLRs that possess a PYD form the NLRP (also known as NALP) subfamily including NALP3 (see Chap. 17). NLRP3 is essentially involved in the activation of the inflammasome , whereas NOD2 plays a role in the activation of NF-κB and mitogen-activated protein (MAP) kinase pathways. Using a variety of cell types including DCs, macrophages, fibroblasts, and melanoma cells, a critical role for the NLRP3 inflammasome has been demonstrated during the infection of various skin-associated viruses, including HSV, VZV, WNV, Japanese encephalitis virus (JEV), measles virus, and HIV-1 (Fig. 19.3) [44, 66–70]. Interestingly, susceptibility to HIV infection in human patients was associated with several polymorphisms in the Nlrp3 gene [71]. Because NALP3 is unlikely to bind directly to viral nucleic acids, NLRP3 inflammasome is considered to be activated through an indirect mechanism. One possibility is that viral RNA is sensed by endosomal TLRs or cytoplasmic RLRs (see Chap. 20.2.4), such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) , leading to the priming of NLRP3 inflammasome for activation and the upregulation of pro-IL–1β through NF-κB–mediated signaling pathways (Fig. 19.3) [72]. In addition, RIG-I has been shown to interact directly with the adaptor protein, apoptosis-associated Speck-like protein containing a CARD (ASC) and regulates inflammasome activation, leading to production of IL-1β. RIG-I can mediate IL-1β production in response to ssRNA virus, vesicular stomatitis virus, by activating the caspase-1–dependent inflammasome, which is required to form a complex containing RIG-I and the adaptor protein ASC in an NLRP3-independent manner [73]. Another group also reported that RIG-I interacts with ASC, and regulates inflammasome activation and NLRP3 transcript levels [74], suggesting that RIG-I may prime and activate the inflammasome via the NLRP3-dependent and -independent pathway (Fig. 19.3). Intriguingly, the encephalomyocarditis virus sensed by MDA did require NALP3 for inflammasome activation [73]. The role of RLRs in inflammasome activation is further complicated because type I IFNs, which can be regulated by RLRs, have also been shown to regulate NLRP3 activation [75].
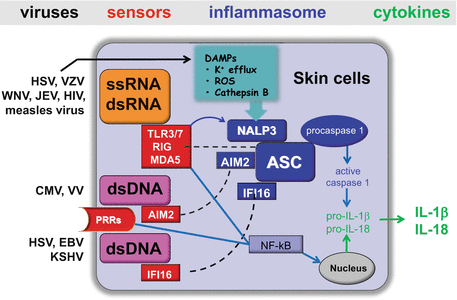
Fig. 19.3
Viral recognition and IL-1β/IL-18 production via inflammasome by skin cells. During the indicated skin-associated virus infections, viral ssRNA or dsRNA is recognized by endosomal TLRs (TLR3/7) or cytoplasmic RLRs (RIG-I or MDA5), leading to the priming of NLRP3 inflammasome for activation and the upregulation of pro-IL–1β/pro-IL–18 through NF-κB–mediated signaling pathways. Following the priming, the NLRP3 inflammasome is assembled and activated in response to virus-induced DAMPs. AIM2 or IFI16 in the cytosol bind viral dsDNA and also engage the adaptor protein ASC to form a caspase-1–activating inflammasome. The AIM2/IFI16 inflammasome requires another signal for IL-1β/IL-18 production, which comes from the virus that triggers other PRR-dependent signaling pathways and allows pro-IL–1β production through NF-κB–mediated signaling
In addition to stimulus from viral ssRNA, dsRNA, and dsDNA, virus-mediated NLRP3 activation requires another signal to induce the production of mature IL-1β by a wide variety of DAMPs including (i) ion leakage from intracellular organelles (e.g., the Golgi) into the cytosol; (ii) the generation of reactive oxygen species resulting from PRR signaling, endoplasmic reticulum (ER), and mitochondrial stress, or virus-induced damage to endosomes, (iii) the P2x7 ion channel is opened in response to extracellular ATP from damaged or necrotic cells allowing for potassium efflux; (iv) during entry, virus infection damages endosomes and releases proteases such as cathepsin B into the cytosol; (v) pore-forming toxins; and (vi) uric acid crystals [36, 72, 76] (See Chap. 17). Indeed, several viral infections have been shown to cause these varieties of DAMPs, which are then recognized by NLRP3, which in turn, recruits the adaptor ASC, and then ASC interacts with the pro-caspsase–1 which becomes activated and allows mature IL-1β production [72].
AIM2 (absent in melanoma 2) has recently been shown to bind DNA and engage the caspase-1–activating adaptor protein ASC to form a caspase-1–activating inflammasome (Fig. 19.3). Knockdown of AIM2 abrogates caspase-1 activation in response to dsDNA of VV and MCMV [77, 78], indicating that AIM2 plays a role in sensing DNA viruses. In addition, another DNA receptor, IFN-γ–inducible protein 16 (IFI16) has been reported to induce ASC-dependent inflammasome activation in a variety of cell types, including DCs, macrophages, and fibroblasts, during infection with DNA viruses such as HSV-1, EBV, and KSHV [77]. NOD2 was also shown to sense several viruses, including Rous sarcoma virus, and mediate host defense via IRF3 and/or mitochondrial antiviral signaling (MAVS) [79]. Recently, it has been shown that the NLR family member NLRX1 interacts with MAVS protein to regulate the interferon and NF-κB pathway negatively [80, 81]. Although conflicting reports have been presented with regard to the implication of NLRX1 as a negative regulator of MAVS-dependent cytosolic antiviral responses [84], NLRs may modulate the host antiviral apparatus either positively or negatively.
19.2.5 RIG-I–Like Receptors (RLRs)
The viral dsRNA in the cytosol can be recognized by an additional family of cytosolic receptors, RLRs, including RIG-I , MDA5 , and LGP2 (laboratory of genetics and physiology 2) (Fig. 19.4) (see Chap. 17) [78, 79]. RLRs are expressed in most skin cell types and are greatly increased with type I IFNs exposure after virus infection. RIG-I and MDA5 contain two CARDs in tandem at their amino terminus that are important for their signaling functions. In general, infection by RNA viruses leads to the generation of long dsRNA in the cytosol that is structurally different from host cellular RNA, which is single-stranded with short and often imperfectly matched stem loops [80]. RIG-I is preferentially involved in the detection of short dsRNA (up to 1 kb) or stem-loop RNA species bearing 5′-triphosphate (5′ ppp) ends, which serve in part to define a nonself RNA PAMP, whereas MDA-5 is more sensitive to long dsRNA (more than 2 kb) containing branched structures [36, 79, 80]. Although RIG-I had initially been implicated in the recognition of ssRNAs with 5′-triphosphate groups, recent studies have demonstrated that pure ssRNA with a 5′-triphosphate group is unable to activate RIG-I [81, 82]. Viral RNA recognition by RIG-I and/or MDA5 leads to the production of type I IFNs via the adaptor protein: mitochondrial antiviral signaling protein (MAVS) and the transcription factors, namely NFκB, IRF3, and IRF7 (Fig. 19.3) [78, 80]. RIG-I and MDA-5 recognize different viruses; RIG-I is essential for immune defense against some skin-associated viruses such as EBV, measles virus, and HIV, whereas MDA-5 is required for IFNs induction by HSV and VV [44, 79]. Some members of the Flaviviridae such as DV, JEV, and WNV have been shown to activate both RIG-I and MDA5 for type I IFNs production in a variety of skin cell types (Fig. 19.4) [79, 83]. It has been demonstrated that during HSV-1 infection, early in vivo production of type I IFNs is mediated through TLR9 in pDCs, whereas the subsequent IFNs response is derived from several cell types and induced independently of TLR9 [84]. In the latter phase, HSV-1 induces type I IFN expression through a mechanism dependent both on RIG-I and the adaptor MAVS [85]. In addition to the RLRs, the helicases DDX1, DDX21, DHX9, and DHX36 have been linked to recognition of cytoplasmic RNA in DCs [44].
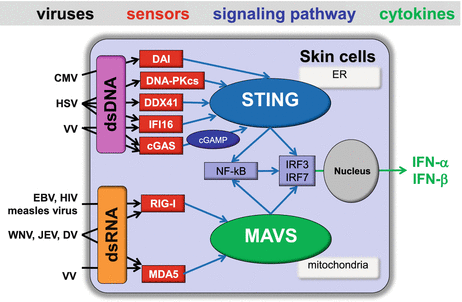
Fig. 19.4
RLRs- and cytosolic DNA sensor-mediated viral recognition and type I IFN production by skin cells. During the indicated skin-associated virus infections, viral dsRNA is recognized by cytoplasmic RLRs (e.g., RIG-I or MDA), leading to the production of type I IFNs via the adaptor protein MAVS and the transcription factors NFκB, IRF3, and IRF7. DNA introduced by invading DNA viruses and/or damaged host cells are released and can meet sensors in the cytosol of skin cells. Multiple DNA sensors, such as DAI, DNA-PKcs, DDX41, cGAS, and IFI16, are considered to activate STING located at the endoplasmic reticulum (ER), culminating in the activation of transcription factors NFκB, IRF3, and IRF7, which then lead to induction of type I IFNs
In general, RNA polymerase III (pol III) has the fundamental role in transcribing transfer RNAs and other small noncoding RNA molecules. Recent studies have demonstrated that pol III is also able to generate dsRNA bearing a 5′ ppp moiety with AT-rich DNA as templates and the dsRNA is then recognized by RIG-I [77, 84]. Although POL3 had initially been implicated in the recognition of HSV-1 and adeno virus [84], other studies have found little evidence for the involvement of this pathway during these viral infections [85, 86]. In EBV infection, pol III play a role in the generation of the EBERs (nonpolyadenylated, noncoding RNA that forms a stem-loop structure by intermolecular base-pairing), giving rise to dsRNA-like molecules, that are subsequently recognized by RIG-I (Fig. 19.4) [84]. However, in this case, pol III acts in its conventional role, that is, in the transcription of functional RNAs, rather than as a sensor per se [80].
19.2.6 Other Intracellular DNA Sensors
One area of recent intense research has been the search for cytosolic DNA-sensing pathways that would account for the ability of exogenously added DNA to induce IFN-β. During viral infection, nucleic acids, such as DNA introduced by invading DNA viruses and/or released from damaged host cells, can meet cytosolic sensors and trigger the activation of innate immune responses, especially when DNA from dead cells is not effectively cleared by DNases, including DNase I in the extracellular space, DNase II in lysosomes, and three prime repair exonuclease 1 (TREX1) in the cytoplasm. In addition to the endosomal TLRs-dependent IFN response elicited by intracellular DNA, it has recently been demonstrated that adaptor protein STING (stimulator of IFN genes, also known as MITA, mediator of IRF3 activation) expressed in the endoplasmic reticulum is also essential for IFN-β induction by intracellular dsDNA and/or DNA viruses via activation of the transcription factors IRF3, TANK-binding kinase 1 (TBK1), and NF-κB [77, 78]. Although the identity of the upstream DNA receptors that cause the activation of STING has remained controversial, it has been proposed that the STING-dependent signaling pathway may be activated by multiple DNA sensors; these include DAI (DNA-dependent activator of IFN-regulatory factors), IFI16, the helicase DDX41, DNA-PKcs (DNA-dependentprotein kinase), and cGAS (cyclic GMP-AMP synthase; Fig. 19.3) [77, 78].
A new cytosolic receptor termed DAI , also known as Z-DNA binding protein (ZBP-1), was found to bind multiple types of exogenously added B-form dsDNA, but not Z-form dsDNA, including viral DNA, leading to the activation of IRF3 which participates in the induction of IFN-β expression in a IRF3- and NF-κB–dependent pathway [87–90]. DAI has been shown to play a role in human fibroblasts during HCMV infection, and another DNA receptor, IFI16 (See Capture 20.2.1.4), DDX41, and the catalytic subunit of DNA-dependentprotein kinase (DNA-PKcs), induce IFN-β in response to intracellular DNA in a variety of skin cell types during HSV-1 infection (Fig. 19.3) [44, 77]. It is of note that IFI16 shuttles between the nucleus and the cytoplasm and senses DNA in both compartments [77]. Several groups have demonstrated that DNA damage can result in the production of type I IFNs [91, 92], suggesting that there are links between viral infection, DNA damage, and the innate immune response. Recent study has shown that DNA damage factors, DNA-PKcs and its binding partners Ku70/80, are required for the production of IFN-β and were also involved in the response to HSV-1 [93]. Although another well-characterized DNA damage factor, Mre11 (Meiotic recombination 11), has also been shown to be required for the DNA-induced IFN response in mouse bone-marrow–derived DCs, Mre11 was not involved in the recognition of pathogens such as HSV-1 [94]. A newly identified enzyme, cGAS , binds intracellular DNA and mediates the production of cyclic GMP-AMP (cGAMP), which functions as a second messenger that binds and activates STING [78]. Thus, cGAS acts as a DNA receptor, and indeed, knockdown of cGAS inhibits IFN-β induction in THP-1 macrophages or the L929 fibroblastic cell line by DNA viruses such as HSV-1 and VV [77, 78].
During HSV-1 and HIV-1 infection, the viral DNA can be recognized by another sensor, high mobility group box 1 (HMGB1) , a DNA-binding nuclear protein [91]. HMGB1 may influence HIV infection at three levels at least; HMGB1 can bind DNA and RNA with high affinity, and promote the activation of TLRs, RIG-I, and intracellular DNA receptors in response to their nucleic acid ligands [92]. HMGB1 have recently been identified to be released outside the cell, and act as a potent soluble factor that coordinates cellular events that are crucial for the amplification of the inflammation and establishment of early innate immune response [91, 92]. HMGB1 is therefore considered as the leading member of the DAMPs, named “alarmins”. HMGB1 is a nuclear protein and, to function as DAMP in the extracellular milieu, it must be released. This occurs either via passive release from necrotic cells, or by active secretion by cells of the innate immune system [92, 93]. Interestingly, extracellular HMGB1 has been shown to inhibit HIV-1 de novo infection, but increases HIV-1 production by persistently infected cells [92].
19.2.7 Antimicrobial Peptides (AMPs)
Virtually all human tissues and cells typically exposed to microbes are able to produce small antimicrobial peptides (AMPs). Defensins and cathelicidins are AMPs that are produced by leukocytes and epithelial cells, and that have an important role in antiviral innate immunity. In addition to their antimicrobial activity, the AMPs also play a role in chemoattraction of lymphocytes and participate in cell growth regulation and wound healing (see Chap. 17). In contrast, CCL20, a representing well-known chemokine, was shown also to be a potent AMP [94]. The known cutaneous AMPs can identify more than 20 individual proteins that have shown antimicrobial activity, including dermcidin, which is constitutively secreted by eccrine sweat glands and psoriasin which was first isolated from psoriatic epidermis, are also the important effector molecules of cutaneous innate immunity (see Chap. 17).
Recent advances in understanding the mechanisms of the antiviral actions of AMPs indicate that they have a dual role in antiviral defense, acting directly on the virion and on the host cell [95]. Human α-defensin-1, -2, -3, and -4 are designated as human neutrophil peptides (HNP1, HNP2, HNP3, and HNP4) because they are mainly expressed by neutrophils, whereas human epidermal keratinocytes or genital epithelial cells can produce human α defensin-5 (HD5), HD6, and human β defensin -1 (HBD1), HBD2, HBD3, HBD4, and the sole cathelicidin in humans, LL-37. Certain defensins (e.g., HBD1) are expressed constitutively, and others (e.g., HBD2 and 3) show increased expression in response to inflammation or infection. For example, HIV-1 and human rhinovirus induce mRNA expression of HBD2 and HBD3 in normal human oral and bronchial epithelial cells, respectively [96, 97]. Since similar upregulation of HBD gene expression is induced by polyinosinic-polycytidylic acid (polyI:C), a ligand for TLR3 [102], TLR3-mediated recognition of dsRNA generated from the viruses might be involved in the upregulation of the HBDs expression. In addition, stimulation of TLR2 and TLR4 has been shown to induce HBD2 expression by vaginal epithelial cells [103].
An emerging body of evidence now indicates the antiviral actions of AMPs. For example, AMPs such as HNP1-4, HBD2, and HBD3 have been reported to inhibit HIV-1 infectivity [95, 98]. Furthermore, α-defensin HNPs (e.g., HNP1, HNP2, HNP3, and HNP4) have inhibitory effects on the infectivity of various skin-associated viruses including HSV-1, HSV-2, and VV. Similarly, cathelicidins and β-defensins, HBD1 and HBD2, also have an inhibitory effect on the infectivity of VV [95]. Cathelicidins are highly expressed by neutrophils and by epidermal or genital epithelial cells, and recent work identified vitamin D3 as a major factor involved in its regulation. Its peptide form, LL-37, was shown to influence TLR signaling in immune cells through interaction with the cellular membrane and epidermal growth factor receptor transactivation and to increase intracellular Ca2+ mobilization [99].
The mechanisms of antiviral action by AMPs are multiple and complex and include direct effects on the virion, as well as effects on the target cell and on innate and adaptive immunity [95]. Recently, we found that HSV-2 significantly increased the expression of hBD2, hBD3, hBD4, and LL-37 in normal human keratinocytes, and surprisingly, LL-37 strongly upregulated the expression of HIV receptors in LCs thereby enhancing their HIV susceptibility [30]. These results may explain the in vivo observation that HSV-2 infection facilitates HIV-1 acquisition. Thus, as many PRRs do, AMPs not only act as beneficial to the host, but also contribute viral infectivity and pathogenicity.
19.3 Intrinsic Antiviral Immunity in Skin
The intrinsic antiviral immunity provides an immediate and direct antiviral defense mediated by intrinsic restriction factors that are mostly pre-existent in certain cell types. Unlike TLRs and RLRs, which inhibit viral infection indirectly by activating signaling cascades that result in the transcription of genes encoding antiviral factors such as type I IFNs (Figs. 19.2 and 19.4), intrinsic restriction factors bind viral components and directly inhibit all steps of viral replication (Fig. 19.5) [100]. Note that as most of these factors are encoded by ISGs, they can be further induced by type I IFNs to amplify their antiviral activity [80].
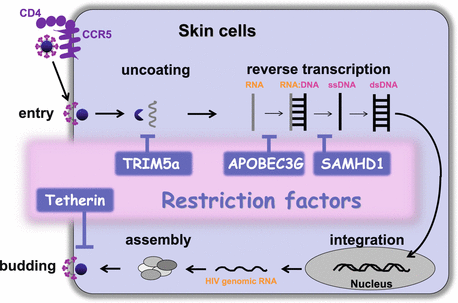
Fig. 19.5
Potential impact of intrinsic antiviral restriction factors on HIV-1 cycle. Certain skin cell types, especially LCs and DCs, express intrinsic antiviral restriction factors, such as TRIM5α, APOBEC3G, SAMHD1, and Tetherin, capable of binding viral components and directly blocking different stages of HIV replication cycle
HIV-1 can infect various skin cells that express CD4 and CCR5, including LCs, DCs, macrophages, and CD4+ T cells. However, the magnitude of infection with HIV-1 is cell type specific, and CD4+ T cells are more susceptible to infection with HIV-1 than LCs/DCs and macrophages. Recent studies have revealed that the distinct susceptibility to HIV-1 among those cell types is largely attributed to the host intrinsic restriction factors, such as APOBEC3G (apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G) and SAMHD1 (SAM domain- and HD domain-containing protein 1; Fig. 19.4) [80, 100]. APOBEC3G was one of the first intrinsic antiviral factors identified as acting against HIV-1. In cells productively infected with HIV-1, host APOBEC3G is packaged into HIV-1 virions, and in the infection to other target cells, APOBEC3G edits C to U in HIV DNA, which results in a G-to-A mutation in the HIV genome, leading to diminished replication. The deoxynucleoside triphosphate SAMHD1 degrades deoxynucleoside triphosphates (dNTPs), and inhibits HIV-1 reverse transcription by decreasing the level of cellular dNTPs to below the level required for the synthesis of viral DNA. Intriguingly, in DCs, SAMHD1 has an additional role in limiting the induction of type I IFNs in response to HIV-1 replication [101]. The capsid-binding protein TRIM5α (tripartite motif 5α) also acts as an intrinsic restriction factor and binds viral capsid targeting the virus for proteasomal degradation prior to reverse transcription. Human TRIM5α mediates mild restriction of HIV-2, but does not cause a significant inhibition to HIV-1, whereas rhesus TRIM5α restricts HIV-1. Tetherin has also been identified as the restriction factor responsible for blocking the release of nascent HIV-1 particles from infected cells [80, 100]. At the HIV-1 budding site, tetherin incorporates into the cell and virus membranes and prevents efficient viral release by tethering HIV-1 particles to the cell, and consequently, tethered particles are internalized by endocytosis and are subsequently degraded in the endosomes (Fig. 19.4). Tetherin has an additional immunological role: the binding of tetherin to ILT7, a membrane receptor selectively expressed in pDCs, leads to the inhibition of TLR-mediated IFN responses [102].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


