, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
3.1 Acropustulosis of Infancy
3.1.1 Clinical Features
Acropustulosis of infancy typically affects young children, with onset between birth and 1 year of age. It is more common in African American infants [1]. Recurrent crops of pruritic vesiculopustules in an acral distribution are characteristic (Figs. 3.1 and 3.2). Lesions typically last up to 2 weeks, clear, and then recur within a few weeks. Severity of outbreaks tends to diminish over time. Spontaneous resolution generally occurs by 2–3 years of age.
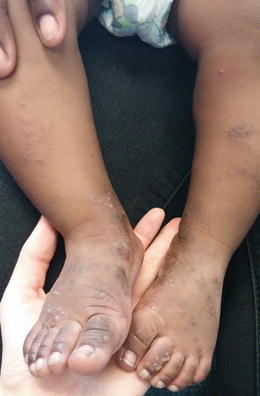
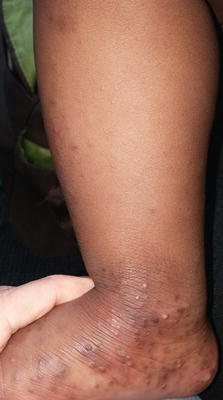
Fig. 3.1
Vesiculopapules and pustules as well as hyperpigmented macules at sites of previously active lesions on the distal legs and feet in acropustulosis of infancy
Fig. 3.2
Acropustulosis of infancy, closer image of the same patient in Fig 3.1
3.1.2 Histology
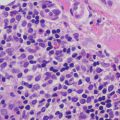
The histologic findings of acropustulosis of infancy are not specific, but characteristic of this entity. The epidermis has foci of spongiosis with microvesiculation [2] (Fig. 3.3). Neutrophilic abscesses are present within the vesicles (Fig. 3.4). Acute lesions do not have parakeratosis, although this is a common finding in more established lesions. Less commonly, eosinophils may be present within the microvesicles [3].
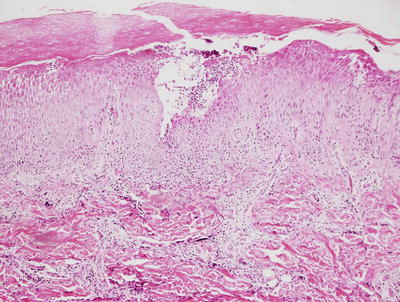
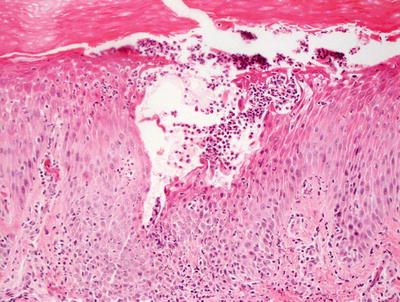
Fig. 3.3
Histologic features of acropustulosis of infancy include intraepidermal spongiosis and clusters of neutrophils within the stratum corneum
Fig. 3.4
Subcorneal neutrophilic clusters are present in acropustulosis of infancy
The differential diagnosis includes candidiasis and impetigo, each of which can be further delineated with use of special stains and microbiological tissue cultures. Pustular psoriasis may present a more difficult diagnostic dilemma in that the usual changes of plaque psoriasis are often not present in acute pustular lesions. The presence of ectatic vessels within the papillary dermis may be some evidence in favor of psoriasis, but in most cases clinical correlation is the best way to make this distinction. In rare cases with eosinophils in the inflammatory infiltrate, erythema toxicum neonatorum enters the differential diagnosis, but it should be easily distinguished based upon the clinical presentation. Similarly, incontinentia pigmenti might show similar histologic features. The presence of dyskeratotic cells favors the diagnosis of incontinentia pigmenti, and the clinical appearance makes for a relatively straightforward distinction.
3.2 Transient Neonatal Pustular Melanosis
3.2.1 Clinical Features
The overall incidence of transient neonatal pustular melanosis is estimated at 0.2–0.6 % [8]. It is more common in African American infants, where incidence is 4.4 % [8]. The disorder is characterized by vesiculopustules on a non-erythematous background. Lesions are typically present at birth or within the first few days of life. Pustules tend to rupture easily leaving hyperpigmented macules behind. These macules usually fade within a few weeks. Although lesions may appear anywhere (including the palms and soles), they tend to favor the chin, neck, upper chest, lower back, buttocks, and thighs [8]. Pustules resolve spontaneously within approximately 1 week. Residual hyperpigmented macules resolve over a few weeks.
3.2.2 Histology
Histologic features include neutrophilic abscesses within the stratum corneum and in the upper portions of the epidermis (Fig. 3.5). Dermal melanin appears in later lesions secondary to post-inflammatory pigmentation. Special stains for fungi and bacteria are negative but are usually required to exclude an infectious etiology. There is only a minimal inflammatory infiltrate within the dermis [9, 10].
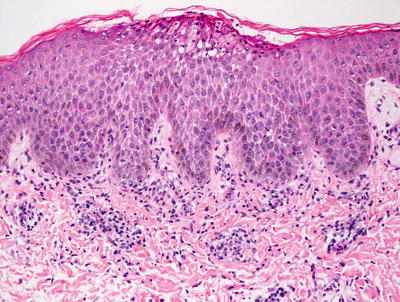
Fig. 3.5
A subcorneal neutrophilic abscess is seen with surrounding spongiosis in transient neonatal pustular melanosis. The basal layer of the epidermis is generally pigmented in patients with this disorder
The histologic differential diagnosis includes pustular psoriasis. This is quite unusual in neonates and can usually be distinguished based upon clinical findings. Vascular ectasia that characterizes psoriasis may be of some use in making this distinction histologically. Subcorneal pustulosis of Sneddon–Wilkinson is also extremely rare in neonates. Erythema toxicum neonatorum occurs in the same population. Histologically, this entity has eosinophils in the pustules in contrast to the neutrophils present in transient neonatal pustular melanosis. This difference allows some authors to consider these two entities to be unrelated [10, 11]. Other authors believe transient neonatal pustular melanosis represents precursor lesions to what rapidly evolves into erythema toxicum neonatorum [9, 12].
3.3 Staphylococcal Scalded Skin Syndrome
3.3.1 Clinical Features
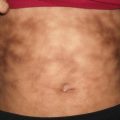
Staphylococcal scalded skin syndrome affects primarily infants and young children, although it has been reported in adults [13]. The primary infection typically involves the conjunctivae, nares, perioral region, perineum and umbilicus [14].
Erythema of the skin progresses to superficial fragile blisters that rupture easily, leaving tender eroded areas behind (Fig. 3.6). Involvement is often most prominent in the flexural creases. Patients may be mildly or severely affected. With extensive desquamation of the skin, there are secondary risks of fluid and electrolyte loss, thermoregulatory instability, and infection. With proper management, the skin is expected to heal without permanent scarring.
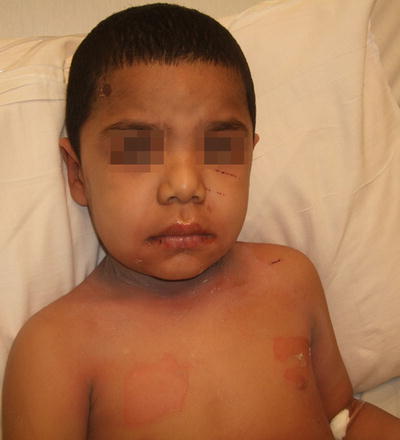
Fig. 3.6
Staphylococcal scalded skin syndrome often presents as tender, dusky red patches at the intertriginous skin with flaccid bullae and superficial erosions on the trunk
3.3.2 Histology
Histologic changes consist of a non-inflammatory subcorneal blister at the level of the granular layer (Fig. 3.7). In many cases, the stratum corneum, which is the blister roof, is desquamated and not present in the biopsy specimen. Its absence is often overlooked, making the diagnosis difficult. Rare acantholytic cells may be observed within the granular layer (Fig. 3.8). There is virtually no underlying inflammation within the dermis. Special stains for bacteria are negative [15].
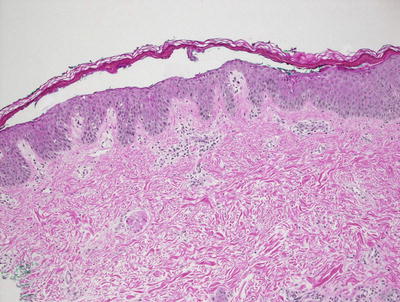
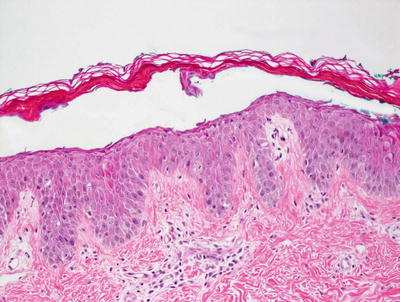
Fig. 3.7
Staphylococcal scalded skin syndrome features a subcorneal blister in the absence of any inflammatory infiltrate
Fig. 3.8
Rare acantholytic cells can be seen at the base of the separation in Staphylococcal scalded skin syndrome
The major differential diagnosis based on histologic grounds is a superficial variant of pemphigus such as pemphigus foliaceus or pemphigus erythematosus . However, these entities have markedly different clinical presentations and are not common in children. Frequently, the pathologist is asked to differentiate staphylococcal scalded skin syndrome from toxic epidermal necrolysis. The histologic distinction is made based upon the level of the blister. In toxic epidermal necrolysis, the blister occurs at the dermal epidermal junction. Abundant dying keratinocytes are present throughout the epidermis, all of which is on the roof of the blister. In contrast, in staphylococcal scalded skin syndrome, all nucleated keratinocytes are on the floor of the blister, with the roof comprised solely of stratum corneum. Impetigo, when caused by staphylococcus, is characterized by neutrophilic abscesses and dermal inflammatory infiltrate. Tissue gram stains demonstrate abundant gram-positive cocci.
3.3.3 Pathogenesis
Staphylococcal scalded skin syndrome is caused by exfoliative toxin produced by Staphylococcus aureus [16–21]. About 5 % of S. aureus human isolates produce exfoliative toxins A and B that cause the disease. Exfoliative toxin disseminates throughout the circulation from a localized site of infection and causes widespread skin involvement at distant sites. Therefore, microbiological cultures of the bullous material in staphylococcal scalded skin syndrome lesions are often sterile.
S. aureus exfoliative toxin is a trypsin-like serine protease [19]. As it accumulates in the skin, the toxin digests desmoglein-1, which is a component of desmosome adhesion between keratinocytes [22–24]. Loss of desmoglein-1 disrupts keratinocyte-to-keratinocyte adhesion in the stratum granulosum, resulting in bullae formation [22, 25]. The relative amount of desmoglein-1 in the skin differs with age group and location in the epidermis. This may partially explain the increased frequency of staphylococcal scalded skin syndrome in young children, and the preferential involvement of the upper layer of the epidermis.
3.4 Subcorneal Pustular Dermatosis of Sneddon–Wilkinson/IgA Pemphigus
3.4.1 Clinical Features
Subcorneal pustular dermatosis is most common in 40–50-year-old women [26]. It is rare in children, although it has been reported in a 7-week-old infant [14]. Subcorneal pustular dermatosis begins with small vesicles or pustules on a normal to slightly erythematous base. Patients experience mild pruritus. The pustules tend to coalesce, forming annular and serpiginous patterns (Fig. 3.9). Lesions tend to occur on the trunk, intertriginous, and flexural areas. The face and mucous membranes are spared. Lesions can rupture, leaving superficial erosions and crusts that heal with blotchy hyperpigmentation. Although benign, subcorneal pustular dermatosis is characterized by remissions and exacerbations that may last for several years. In adults, patients require long-term follow-up, as there is a reported association with possible development of monoclonal gammopathy and multiple myeloma.
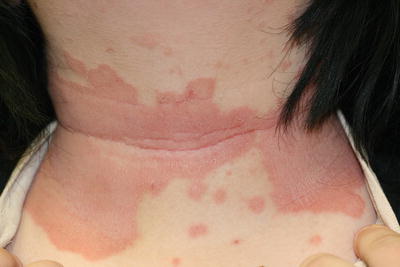
Fig. 3.9
Subcorneal pustular dermatosis of Sneddon–Wilkinson (IgA pemphigus) presents as erythematous, serpiginous plaques at the anterior neck and superior trunk
3.4.2 Histology
IgA pemphigus, likely identical to Sneddon–Wilkinson syndrome , has two predominant histologic patterns. In one pattern, neutrophils aggregate as subcorneal pustules, while in the other, they are deeper within the epidermis and accompanied by slight acantholysis [27]. Even less commonly, epidermal acanthosis resulting in a pattern resembling pemphigus vegetans has been reported [28]. Direct immunofluorescence examination reveals intercellular staining throughout the epidermis with anti-IgA antibodies (Fig. 3.10).
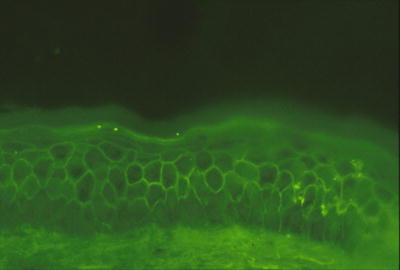
Fig. 3.10
Intercellular staining throughout the epidermis with anti-IgA antibodies characterizes IgA pemphigus
The histologic differential diagnosis is dependent upon the histologic pattern. In the subcorneal form of the disease, a wide range of subcorneal blistering processes occurring in the pediatric population must be considered. Special stains can help to exclude the infectious processes such as candidiasis and impetigo. Clinical history might help with transient neonatal pustular melanosis and acral pustulosis of infancy. The variant with intraepidermal abscesses must be distinguished from infectious etiologies. As with all of the pemphigus subtypes, resolution of the differential diagnosis is simplified by direct immunofluorescence stains. Other types of pemphigus are quite rare in children. Pemphigus foliaceus , demonstrating blister formation within the granular layer of the epidermis, has been described in children [29].
3.4.3 Pathogenesis
The etiology of subcorneal pustular dermatosis is not known. Infection does not appear to be a cause, since microbiological cultures of the pustules in this disorder do not show growth of microorganisms [30]. Subcorneal pustular dermatosis may have immunologic mechanisms. Some patients with the disorder have circulating IgA antibodies to desmocollins-1, 2, and 3, which have been described as auto-antigens in some cases [31–33]. However, the exact role of these antibodies in the pathogenesis of the disease remains to be determined.
3.5 Acute Generalized Exanthematous Pustulosis
3.5.1 Clinical Features
Acute generalized exanthematous pustulosis (AGEP) is most common in adults, although it has been reported in some children [34]. Mortality rate is 2 %. AGEP is characterized by the acute onset of an eruption composed of generalized erythroderma and non-follicular, sterile pustules (Fig. 3.11). This typically occurs in the setting of fever. It generally resolves spontaneously with patchy superficial desquamation within 4–10 days. Recurrences are expected to occur with reexposure to the offending trigger and are typically faster in onset.
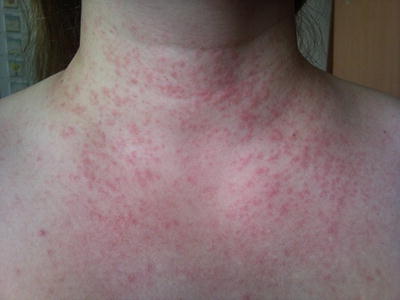
Fig. 3.11
Generalized, non-follicular based erythematous papules and pustules as seen on the trunk of an adolescent patient with AGEP (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
3.5.2 Histology
AGEP is uncommon in children, but when it occurs, shows the same histologic changes as have been reported in adults [35]. Subcorneal pustules comprised of neutrophils are present within a slightly spongiotic epidermis (Fig. 3.12). Parakeratosis is infrequent, except in older lesions. The granular layer persists and acanthosis is not a common finding. Within the dermis, there is a mixed inflammatory infiltrate that consists of neutrophils, eosinophils, and histiocytes [36] (Fig. 3.13).
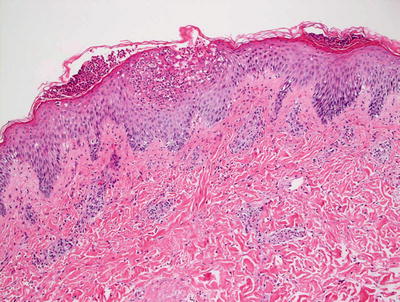
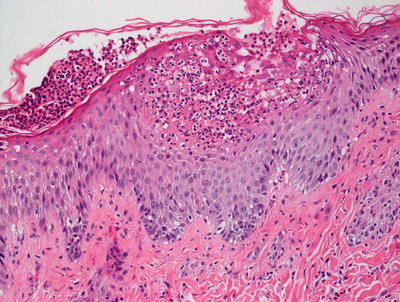
Fig. 3.12
AGEP demonstrates subcorneal and intraepidermal neutrophilic abscesses. There is focal spongiosis and a mild, mixed superficial dermal perivascular infiltrate
Fig. 3.13
In addition to the neutrophilic abscesses in the epidermis, AGEP has neutrophils and eosinophils within the dermal infiltrate
The major histologic differential diagnosis is pustular psoriasis that can be distinguished by the absence of eosinophils, occasional acanthosis and suprapapillary thinning. However, in many acute cases, these changes may not be well developed and distinction may be quite difficult without the aid of clinical history [37]. Infectious etiologies such as impetigo and candidiasis might present some diagnostic difficulties, but this can usually be resolved with the use of special stains for microorganisms. Transient neonatal pustular melanosis can be distinguished most easily based upon clinical presentation. The inflammatory infiltrate in erythema toxicum neonatorum is predominantly eosinophilic with only a minority population of neutrophils unlike in AGEP.
3.5.3 Pathogenesis
AGEP is caused by drugs in most cases. Common drugs associated with AGEP are pristinamycin, aminopenicillins, quinolones, hydroxychloroquine, sulfonamides, terbinafine, and diltiazem [38–40]. Infectious agents, such as coxsackie virus , cytomegalovirus , parvovirus B19 , Chlamydia pneumoniae , and Escherichia coli , are also associated with this disorder [41].
AGEP may have a genetic basis. Mutation frequency in IL-36 receptor antagonist (IL36RN) is higher in patients with AGEP than in control population (1.6 % vs. 0.4 %) [42]. It has been suggested that IL36RN gene mutation may be a predisposing genetic factor to developing AGEP.
AGEP is a T lymphocyte-mediated reaction. Exposure to the causative agent results in the presentation of the inciting antigen by antigen-presenting cells to T lymphocytes . Activation, proliferation and migration of drug-specific CD4+ and CD8+ T lymphocytes are observed in AGEP [40, 43]. Infiltrating CD4+ T lymphocytes and keratinocytes release CXCL-8 (a potent neutrophilic cytokine) and granulocyte macrophage-colony stimulating factor (GM-CSF), which result in the recruitment of neutrophils and the formation of neutrophilic pustules [43]. Drug-specific CD4+ T lymphocytes from patients with AGEP are predominantly type 1 T helper cells (Th1) with Th1-type cytokine profile and the production of interferon-γ (IFN-γ) and GM-CSF [44, 45]. IFN-γ and GM-CSF augment neutrophil survival. There is also increased IL-15 production, which is a potent stimulator of eosinophil proliferation and differentiation. Type 17 T helper cells (Th17) also play a role in AGEP [46, 47]. Th17 cells produce IL-17 and IL-22, which increase the production of CXCL8 by keratinocytes. Apoptosis of keratinocytes is mediated by CD8+ cytotoxic T lymphocytes via perforin/granzyme B and Fas ligand mechanisms, leading to epidermal necrosis and vesicle formation as observed in this disorder [43, 48].
3.6 Friction Blister
3.6.1 Clinical Features
Friction blisters are very common, especially in active individuals. Blisters result from frictional forces that mechanically induce cleavage of the epidermis at the level of the stratum spinosum. The area of separation fills with fluid secondary to hydrostatic pressure. Blisters are more common in areas with a thick horny layer that is tightly adherent to underlying tissues (e.g., palms and soles) [49]. If the bulla is de-roofed, there is a tender erosion. With adequate wound care and protection from additional trauma, friction blisters typically heal spontaneously within several days.
3.6.2 Histology
Blisters caused by friction can occur at any level of the epidermis or commonly at the dermal-epidermal junction. The blisters are non-inflammatory unless there is secondary infection and inflammation as a reaction to the injury (Fig. 3.14). Epidermal necrosis is only present after a period of time during which the keratinocytes on the roof of the blister are devoid of blood flow.
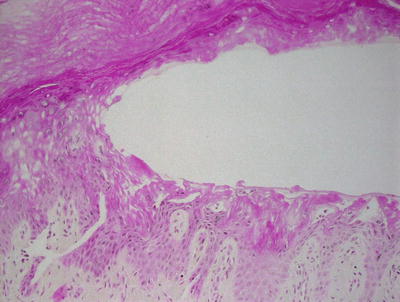
Fig. 3.14
A mid-epidermal blister without any immune response is characteristic of a friction blister
The histologic differential diagnos is is small. Epidermolysis bullosa is the closest histologic mimic and in fact, this family of diseases results in mechanically induced blisters identical to friction blisters. However, clinical history easily resolves the diagnostic dilemma.
3.7 Erythema Toxicum Neonatorum
3.7.1 Clinical Features
Erythema toxicum neonatorum is a benign skin eruption of the newborn that typically appears 24–48 hours after birth. Up to 70 % of newborns are affected. Although it does not appear to have a gender or racial predilection, it is more common with increasing gestational age and birth weight.
Clinically, lesions appear as small white papules or pustules on an erythematous base (similar to a “flea bite”). They are usually most numerous on the trunk, but may also occur on the face, arms, and legs. Palms and soles are typically not affected. Lesions clear within 5–7 days without discoloration or permanent scarring.
3.7.2 Histology
Histologic sections demonstrate skin with subcorneal abscesses containing a predominance of eosinophils (Fig. 3.15). Parakeratosis is variably present depending upon the age of the lesion. Within the dermis, there is a relatively slight inflammatory infiltrate that consists of lymphocytes, histiocytes, and scattered eosinophils. Increased numbers of Langerhans cells are also present within lesions [50].
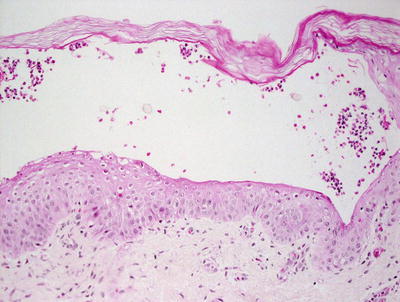
Fig. 3.15
Erythema toxicum neonatorum shows a subcorneal eosinophilic abscess
The differential diagnosis includes incontinentia pigmenti, which is differentiated based upon the presence of dying keratinocytes and is not a feature of erythema toxicum neonatorum. Eosinophilic abscesses may also be encountered in IgA pemphigus, although the clinical history and age of the patient will help to make this distinction. Other subcorneal pustular processes are easily distinguished by the predominance of neutrophils as opposed to the eosinophils that characterize erythema toxicum neonatorum. Some authors have suggested that neutrophilic abscesses may be present in erythema toxicum neonatorum and that differentiation from transient neonatal pustular melanosis is not always possible [12].
3.8 Hydroa Vacciniforme
3.8.1 Clinical Features
Hydroa vacciniforme is common in children with disease onset typically between the ages of 1 and 15 years old. Prevalence of hydroa vacciniforme has been reported at 0.34 cases per 100,000 individuals [51]. Hydroa vacciniforme is a photodermatosis. The primary skin lesions are edematous papules, vesicles or bullae that appear within hours to days after sun exposure. Lesions tend to favor exposed areas, such as the face, neck, and extensor arms and hands. Patients report itching and burning of the skin within hours of sun exposure, and may also have mild constitutional symptoms. Vesicles and bullae tend to crust over within a few days and heal with varioliform scarring (Fig. 3.16). The course of hydroa vacciniforme tends to be relapsing and remitting, although it typically improves or resolves after puberty [14]. Children with hydroa vacciniforme in the setting of chronic Epstein–Barr virus infection appear to have an increased risk of lymphoproliferative disease and should be monitored closely [51].
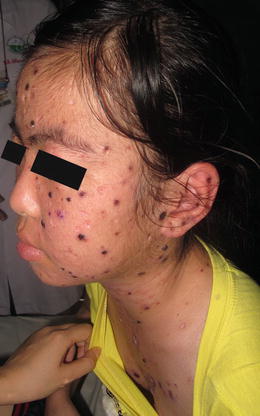
Fig. 3.16
Hydroa vacciniforme presents as photodistributed, crusted, erythematous papules and vesicles (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
3.8.2 Histology
The histologic findings in hydroa vacciniforme are not specific. The epidermis is markedly spongiotic in early lesions, progressing to reticular degeneration and ultimately necrosis in later stage lesions (Fig. 3.17). Within the dermis, there is a brisk infiltrate that consists predominantly of lymphocytes and histiocytes (Figs. 3.18 and 3.19). Eosinophils are not abundant. Neutrophils may be observed in ulcerated lesions, but this is likely a secondary phenomenon.
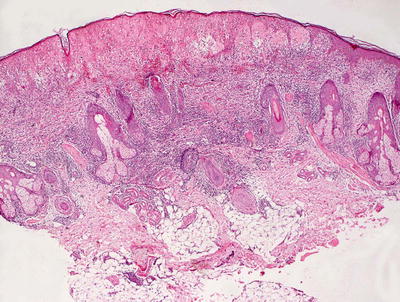
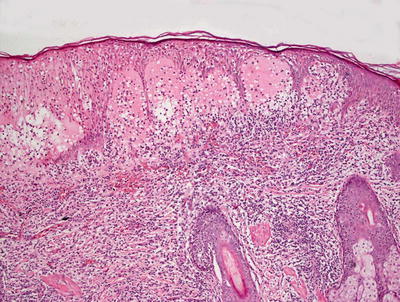
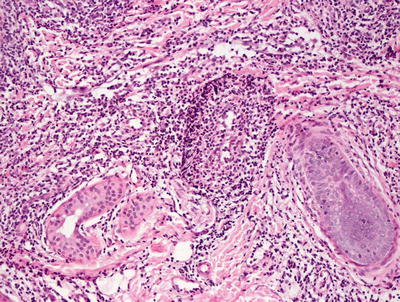
Fig. 3.17
Hydroa vacciniforme demonstrates a subepidermal blister with abundant dermal edema and a brisk inflammatory infiltrate
Fig. 3.18
A brisk inflammatory infiltrate in the dermis extends into the overlying epidermis in hydroa vacciniforme. Blister formation is due to spongiosis and can be at any level
Fig. 3.19
Hydroa vacciniforme is characterized by a brisk superficial and often deep dermal inflammatory infiltrate
The histologic differential diagnosis in early lesions or mild cases includes spongiotic processes, such as atopic dermatitis and nummular eczema. As the degree of epidermal edema increases, other entities, such as polymorphous light eruption and herpetic dermatitis, enter the differential diagnosis. Polymorphous light eruption does not ordinarily result in epidermal necrosis, and the inflammatory infiltrate is often deeper and not quite as intense as that seen in hydroa vacciniforme. The marked papillary dermal edema seen in some cases of polymorphous light eruption might also help with this distinction. Herpetic infection demonstrates the same reticular degeneration that characterizes florid cases of hydroa vacciniforme, but keratinocyte multinucleation, nuclear molding, and viral inclusions seen with herpes infections are not present in hydroa vacciniforme. There have been several reports of cutaneous T cell and natural killer cell lymphomas that have similar clinical findings [52–55]. In these cases, the cytologic features of the inflammatory infiltrate should help to elucidate the true nature of the process.
3.8.3 Pathogenesis
The cause of hydroa vacciniforme is not known. However, ultraviolet A (UVA) appears to have an important role in the disease [56–58]. Lesions characteristic of hydroa vacciniforme can be reproduced with artificial UVA exposure. Infections with Epstein–Barr virus (EBV) may also be causative. Increased levels of EBV DNA have been found in the peripheral blood of patients, and EBV-encoded small nuclear ribonucleic acid (EBER) is present in skin lesions in classic hydroa vacciniforme and hydroa vacciniforme-like cutaneous eruptions [59–61].
3.9 Hailey–Hailey Disease
3.9.1 Clinical Features
Skin manifestations of Hailey–Hailey disease usually begin in the late teens or early twenties [14]. Hailey–Hailey disease is characterized by vesiculobullous lesions, crusted erosions, and vegetative plaques, occurring mainly at the neck, axillae, groin, and perineum (Fig. 3.20). Patients may have significant burning, pruritus, and tenderness in the affected areas. The course of Hailey–Hailey disease is chronic and unremitting. Heat, humidity, friction, and secondary infection tend to exacerbate the disease [62].
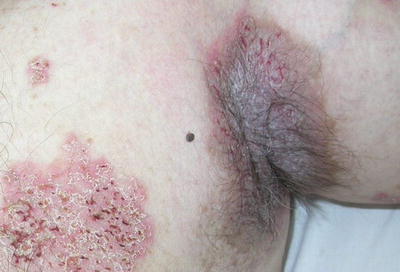
Fig. 3.20
Crusted and eroded vegetative plaques at the axilla and anterior trunk characterize Hailey–Hailey disease (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
3.9.2 Histology
Histologic features of Hailey–Hailey disease show an epidermis with varying degrees of spongiosis and acantholysis. The stratum corneum has normal thickness. The roof of the intraepidermal blister is often fragmented with single keratinocytes separating from other keratinocytes, the so-called “dilapidated brick wall” appearance (Figs. 3.21 and 3.22). Dyskeratotic cells are not abundant. An inflammatory infiltrate that is predominantly lymphohistiocytic with scattered eosinophils is present and may extend into the epidermis [63, 64].
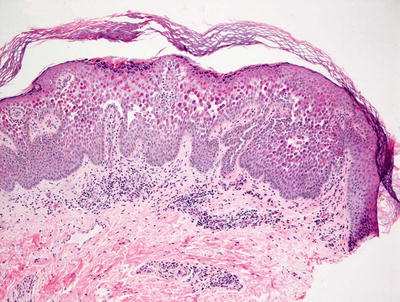
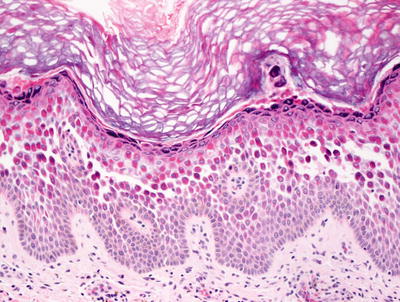
Fig. 3.21
Hailey–Hailey disease shows a mid-epidermal or suprabasilar blistering process within a spongiotic epidermis
Fig. 3.22
The roof of the blister in Hailey–Hailey disease appears to be disintegrating as individual keratinocytes separate into the blister cavity from above
The histologic differential diagnosis includes other vesiculobullous disorders, such as Darier’s disease and Grover’s disease. Darier’s disease is easily distinguished based upon the presence of marked hyperkeratosis, acanthosis, corps ronds, and grains. Grover’s disease may display an identical histologic appearance but the disorder is usually not seen in the pediatric population. Herpetic dermatitis may also represent a diagnostic dilemma, but the presence of viral changes, as well as the clinical presentation, easily distinguishes these diseases from Hailey–Hailey disease . A florid spongiotic dermatitis might enter into the differential diagnosis. The clinical history may be the best way to make the distinction. While intertrigo may demonstrate spongiosis and share some clinical features, the histologic changes are quite mild in intertrigo when compared with the changes seen in Hailey–Hailey disease.
3.9.3 Pathogenesis
Hailey–Hailey disease is caused by mutations in the ATP2C1 (ATPase, Ca2+ transporting, type 2C, member 1) gene, which encodes the secretory pathway Ca2+/Mn2 ATPase (hSPCA1) [65, 66]. Over 100 different mutations in ATP2C1 have been found in patients with Hailey–Hailey disease, many of which lead to haploinsufficiency in the protein product hSPCA1 [67, 68]. hSPCA1 is a magnesium-dependent enzyme that catalyzes the hydrolysis of ATP coupled with the transport of calcium ions [69]. hSPCA1 functions as a calcium pump that regulates the sequestration of calcium in the Golgi apparatus [70, 71]. Desmosomes and adherens junctions between keratinocytes consist of calcium-binding transmembrane glycoproteins. Perturbations in calcium homeostasis can result in impairment in these junctions, and consequently cause acantholysis of the epidermis. The calcium content of basal keratinocytes in Hailey–Hailey disease is lower than that in normal skin, consistent with perturbations in calcium regulation in the disease [65, 68, 72].
3.10 Darier Disease
3.10.1 Clinical Features
Classic skin findings of Darier disease typically appear between 8 and 15 years of age [14]. The disease prevalence is estimated at 1 in 50,000 individuals [73]. Males and females are equally affected. Skin lesions are hyperkeratotic papules with waxy scale occur mainly in a “seborrheic” distribution, favoring the scalp, forehead, ears, nasolabial creases, upper chest, and upper back. Papules coalesce into crusted plaques that often become malodorous. Punctate keratoses and pits may occur on the palms and soles (Figs. 3.23 and 3.24). Thin keratoses resembling flat warts appear on the dorsal hands and are known as acrokeratosis verruciformis . Nails tend to be fragile with characteristic V-shaped nicking at the edges and longitudinal alternating red and white streaks. Darier disease has a chronic and unremitting clinical course. Sun and heat exposure tend to exacerbate the disease.
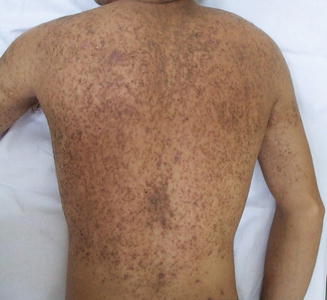
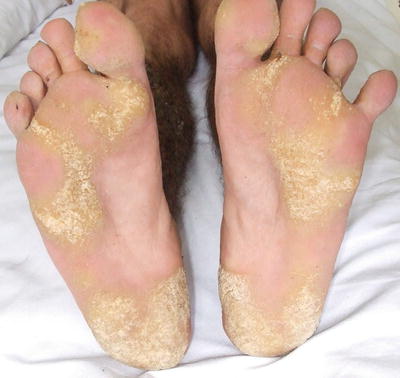
Fig. 3.23
Lesions in Darier’s disease are hyperkeratotic, malodorous, hyperpigmented papules coalescing into plaques in a somewhat seborrheic distribution at the trunk (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
Fig. 3.24
Hyperkeratosis with pitting along pressure points of both soles in Darier disease (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
3.10.2 Histology
Histologic sections show skin with marked hyperkeratosis and focal parakeratosis (Fig. 3.25). The epidermis is acanthotic with elongation of the rete ridges. Dyskeratotic keratinocytes are frequent with corps ronds and grains that are easy to find in most cases (Fig. 3.26). Spongiosis is present focally, but it is not a prominent feature. There is no significant underlying inflammatory response in most cases.
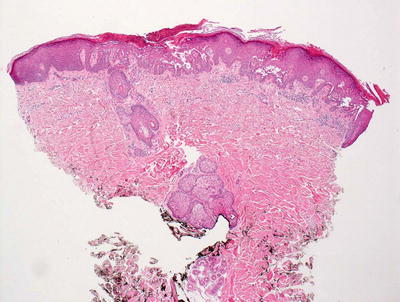
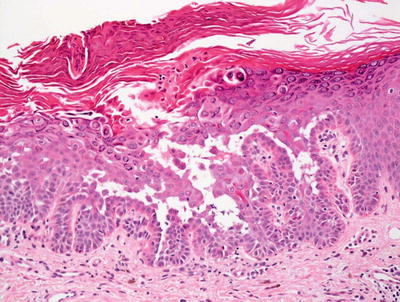
Fig. 3.25
Hyperkeratosis and hypergranulosis are present in Darier disease. Foci of intra-epidermal acantholysis are present resulting in suprabasilar blister formation
Fig. 3.26
Abundant corps ronds and grains are present in the setting of a suprabasilar acantholytic blister in Darier disease
The histologic differential diagnosis includes other acantholytic processes and intraepidermal blistering disorders. Hailey–Hailey disease can be distinguished based upon clinical presentation, as well as histologic differences. Dyskeratosis and hyperkeratosis are not significant features of Hailey–Hailey disease, which is characterized more by extensive spongiosis. Herpetic dermatitis also demonstrates acantholysis, but viral changes such as nuclear molding, multinucleation and nuclear inclusions are also observed in addition to a far more intense inflammatory response in most cases. Pemphigus vulgaris , another acantholytic process, is uncommon in children and does not ordinarily feature prominent dyskeratosis or hyperkeratosis. (The pemphigus vegetans variant may be hyperkeratotic, but has acantholysis without dyskeratosis.) A warty dyskeratoma may demonstrate identical features to Darier disease, although its architecture is that of a cup-shaped lesion. These are extremely uncommon in children. Similarly, dyskeratotic acanthoma and some variants of Grover’s disease (transient acantholytic dermatosis) may appear identical to Darier disease microscopically, but they do not enter into the differential diagnosis in the pediatric population.
3.10.3 Pathogenesis
Darier disease is an autosomal dominant disease with high penetrance (95 %) [74, 75]. The gene implicated in Darier disease is ATP2A2 (ATPase, calcium-transporting, cardiac muscle, slow twitch 2), which encodes SERCA-2 protein [76, 77]. SERCA-2 is a calcium ATPase that transports calcium from the cytosol back to the endoplasmic reticulum (ER) lumen, and facilitates the sequestration of cytosolic calcium in the ER [78–80]. Abnormalities in SERCA-2 can deplete calcium stores in the ER in keratinocytes and disrupt the assembly of the desmosomal complex. In fact, electron microscopic studies have shown loss of desmosomes, breakdown of desmosome–keratin intermediate filament attachment, and perinuclear aggregates of keratin intermediate filaments in Darier disease [81, 82]. Thus, SERCA-2 is important for proper cell-to-cell adhesion. Depletion of calcium ER stores in the cell can have other negative consequences. It can impair posttranslational modifications of newly synthesized proteins and trigger the ER stress response [80, 83]. In fact, the trafficking of desmoplakin, which is a protein component of desmosomes, is defective in Darier keratinocytes [84]. Activation of apoptosis in keratinocytes in Darier disease is another consequence of perturbations in calcium regulation [85].
In Darier disease, there are over 130 mutations that disrupt critical functional domains of SERCA-2 with mutations rendering SERCA2 haploinsufficiency in which mutations in one allele are sufficient to cause the disease phenotype [76, 86, 87]. These mutations lead to a reduction in the expression and activity of SERCA-2 by enhancing proteasome-mediated degradation [88]. There is decreased expression of SERCA2 in the epidermis in Darier disease skin as compared with normal skin [89]. Mosaicism in ATP2A2 mutations can result in segmental Darier disease [90].
3.11 Epidermolysis Bullosa Simplex
3.11.1 Clinical Features
The prevalence of epidermolysis bullosa simplex is estimated at 4.6 per million, and the incidence is approximately 10.8 per million [91]. All forms of epidermolysis bullosa are caused by mutations in a variety of structural genes in the skin, resulting in increased fragility of the skin and blister formation. Most forms of epidermolysis bullosa simplex are inherited in an autosomal dominant manner.
Localized epidermolysis bullosa simplex (the most common variant) typically presents in childhood or adolescence with trauma-induced blisters, primarily located on acral surfaces. As blistering occurs within the epidermis, lesions typically heal rapidly without residual scarring. Nails and mucous membranes are generally not affected. The more severe forms of epidermolysis bullosa simplex, generalized and Dowling–Meara forms, typically present with blistering at birth. They are also unlikely to cause scarring, although patients are more likely to have nail dystrophy and oral involvement. Overall, patients with epidermolysis bullosa simplex have a relatively good prognosis. Many patients show improvement over time. Patients with the less common subtypes of epidermolysis bullosa simplex tend to have more complications and a poorer prognosis.
3.11.2 Histology
The blistering in epidermolysis bullosa simplex occurs within basal keratinocytes. This separation is within the cytoplasm, beneath the nuclei. Thus, the histologic appearance is that of a blister that is above the basement membrane zone, located within the basal layer of the epidermis (Fig. 3.27). In many cases, however, it is difficult to detect the small residual fragments of basal keratinocytes that remain attached to the floor of the blister, giving rise to the appearance of a subepidermal blistering process. The small fragments of basal keratinocytes tend to retract and lie flat along the basement membrane and may not be easily detected on H&E stains [92]. Ultrastructural analysis will easily resolve the level of the blistering (Fig. 3.28) [93]. Since this is a mechanically induced blistering disorder, an inflammatory infiltrate is not ordinarily part of the process, except as a secondary phenomenon that is seen in older lesions. Histologic scarring is not seen in epidermolysis bullosa simplex.
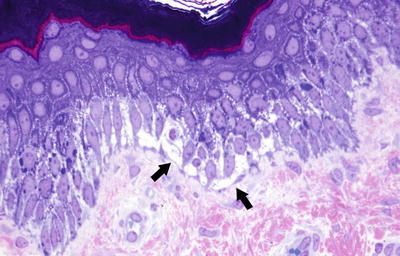
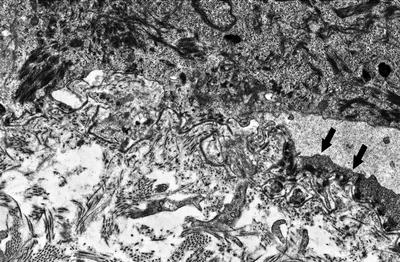
Fig. 3.27
Epidermolysis bullosa simplex has an intraepidermal split within the lower half of basal keratinocytes (photo courtesy of Dr. John Hicks, Texas Children’s Hospital, Houston, TX)
Fig. 3.28
A blister is seen above the basement membrane zone with arrows indicating keratinocyte cytoplasm at the base of the blister cavity in epidermolysis bullosa simplex (photo courtesy of Dr. John Hicks, Texas Children’s Hospital, Houston, TX)
The histologic differential diagnosis includes all cell-poor, non-inflammatory subepidermal blistering disorders, despite the fact that the blister is not actually subepidermal in epidermolysis bullosa simplex. Entities such as porphyria can be differentiated based upon the PAS positive, thick-walled blood vessels in the papillary dermis.
3.11.3 Pathogenesis
There are three major subtypes of epidermolysis bullosa simplex—the severe (Dowling–Meara) type, the moderate and generalized (Koebner) type, and the mild localized (Weber–Cockayne) type [94, 95]. Epidermolysis bullosa simplex is caused by mutations in keratin 5 and keratin 14, resulting in abnormal keratin network in the epidermis and blister formation within the cytoplasm of basal epidermal keratinocytes [96–101]. The position of mutations in keratin 5 and keratin 14 genes correlate closely with the resulting phenotype. For example, the severe (Dowling–Meara) type is often caused by mutations in the initiation or termination peptides of the rod domain of keratin [100, 102]. The mild localized (Weber–Cockayne) type is caused by mutations in other parts of the rod domain or in the linker region of keratin. There are other rare forms of epidermolysis bullosa simplex that are recessively inherited, namely epidermolysis bullosa simplex with muscular dystrophy and pyloric atresia that are caused by mutations in the plectin gene [103–106].
3.12 Epidermolysis Bullosa, Junctional Type
3.12.1 Clinical Features
Junctional epidermolysis bullosa presents in infancy. Blistering is nearly always present at birth. Children typically have generalized blistering and large erosions (Fig. 3.29). Nail dystrophy and nail loss are common. Exuberant perioral granulation tissue is common in the Herlitz subtype. The mucous membranes are frequently affected. The teeth tend to have hypoplastic enamel and are prone to decay. Blisters tend to heal with atrophic scarring. Malnutrition and growth failure may occur.
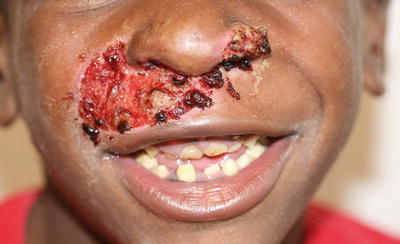
Fig. 3.29
An erosion involving the nasal ala and upper cutaneous lip with distinctive periorifical granulation tissue is seen in a boy with junctional epidermolysis bullosa. Irregular tooth enamel is seen as well
The Herlitz form is associated with high mortality in the first 2 years of life (Fig. 3.30). Death occurs in 40 % of the cases by 1 year of age, and 50 % by 2 years of age [91]. Common causes of death are failure to thrive, sepsis, and respiratory failure. Patients with the non-Herlitz subtype are more likely to survive into adulthood.
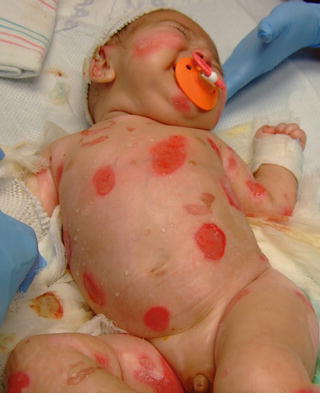
Fig. 3.30
Multiple tense bullae and large erosions in an infant with LAMB-3-associated junctional epidermolysis bullosa (Herlitz variant)
3.12.2 Histology
Histologic changes of junctional epidermolysis bullosa are those of a non-inflammatory subepidermal blister (Fig. 3.31). The epidermis is unremarkable and there is a clean separation from the dermis beneath the basal keratinocytes. Within the underlying dermis, the only inflammatory response is a secondary one in long-standing lesions. There is no keratinocyte necrosis except in older lesions [92]. The absence of necrosis and inflammation helps to distinguish epidermolysis bullosa of the junctional and dystrophic types from other types of subepidermal blistering disorders.
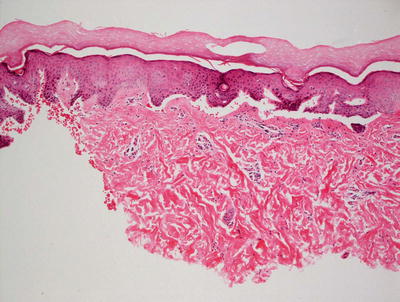
Fig. 3.31
Junctional epidermolysis bullosa is histologically identical to the dystrophic variant, demonstrating a non-inflammatory subepidermal blister
The histologic differential diagnosis includes other non-inflammatory subepidermal blistering processes. The dystrophic variant of epidermolysis bullosa can be distinguished from the junctional variant with the use of immunomapping and ultrastructural studies as well as on their different clinical presentations. The presence of underlying dermal scarring is more apparent in the dystrophic variant than in junctional types of epidermolysis bullosa.
3.12.3 Pathogenesis
Junctional epidermolysis bullosa (JEB) is divided into two main types—the lethal Herlitz type with extreme fragility of the skin and mucous membranes, usually leading to death early in life, and the milder non-Herlitz type [95, 107]. JEB-Herlitz type is caused by loss-of-function mutations in LAMA3, LAMB3, and LAMC2, which are protein components of laminin 332, resulting in the complete loss of laminin 332 [108–111]. Laminin 332 is a major extracellular matrix component in the basement membrane. The absence of laminin 332 causes tissue separation along the lamina lucida of the epidermal basement membrane, so that the intact epidermis separates into the roof of the blister and the basement membrane is on the floor of the blister [112]. The presence of any of the three laminin 332 chains (LAMA3, LAMB3 and LAMC2) can be detected by immunofluorescence stains. Absence of signals for these chains indicates JEB-Herlitz type [108]. LAMB3 is the main gene affected in about 80 % of the cases of JEB-Herlitz, and it is usually the first gene to be screened [113]. The most frequent mutation in LAMB3 is R635X, a mutational hot spot, which accounts for over 60 % of all mutated LAMB3 alleles in JEB-Herlitz [111].
JEB-non Herlitz type is caused by mutations in LAMA3, LAMB3 and LAMC2 that result in reduced amounts of functional laminin 332, but not complete loss of laminin 332 as seen in JEB-Herlitz type [109, 114, 115]. JEB-non Herlitz can also be caused by mutations in COL17A1 gene, which encodes collagen XVII, a binding ligand of laminin 332 [116]. Collagen XVII can be detected by immunofluorescence stains, and absence of collagen XVII signal indicates JEB-non Herlitz type. There are also rare cases of JEB with pyloric atresia that is caused by loss of integrin α6 or β4 subunits, which are the main receptor for laminin 332 [117, 118].
3.13 Epidermolysis Bullosa Dystrophica
3.13.1 Clinical Features
Dystrophic epidermolysis bullosa is divided into two major subtypes, recessive dystrophic epidermolysis (RDEB) and dominant dystrophic epidermolysis bullosa (DDEB) . RDEB has a prevalence of 0.9 per million, and an incidence of 2 per million [91].
In general, patients with DDEB have a relatively mild phenotype due to partial reduction in the expression of collagen VII as compared to the more severe RDEB where patients have a complete absence of collagen VII. Blistering is limited to areas of pressure and friction, such as the hands, feet, elbows, and knees. Nail dystrophy is common, especially the great toenails. Patients with DDEB tend to have a relatively good prognosis, showing improvement in blistering after early childhood.
RDEB is the most severe form of epidermolysis bullosa. Patients tend to have a progressive course with poor quality of life and shortened life span. RDEB patients typically have widespread blistering at birth (Fig. 3.32). Severe mucosal involvement affecting the eyes, the gastrointestinal tract, and the genitourinary tract is common. Wounds are deeper and tend to heal with scarring. Over time, this leads to syndactyly of the fingers and toes. Patients are also susceptible to development of aggressive squamous cell carcinoma, which is the most common cause of early death [91].
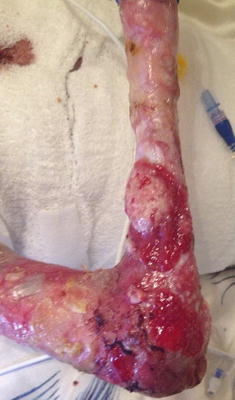
Fig. 3.32
Wide spread erosions and hyperplastic granulation tissue may represent malignancy in later stages of recessive dystrophic epidermolysis bullosa
3.13.2 Histology
The histologic changes in dystrophic epidermolysis bullosa are largely indistinguishable from those seen in the junctional variant. A non-inflammatory subepidermal blister is present with no other alterations in the epidermis [92]. In contrast to the junctional form of epidermolysis bullosa, prominent dermal scarring may be present, especially if the biopsy is taken from an area of repeated blistering. In these cases, there may be a slight inflammatory response within the dermis secondary to repeated ulceration and healing; however, the inflammation is reactive and not part of the primary disease process (Fig. 3.33). Ultrastructural studies demonstrate the presence of blister formation deep to the lamina densa due to reduction (dominant forms) or absence (recessive forms) of anchoring fibrils comprised of type VII collagen [119–122].
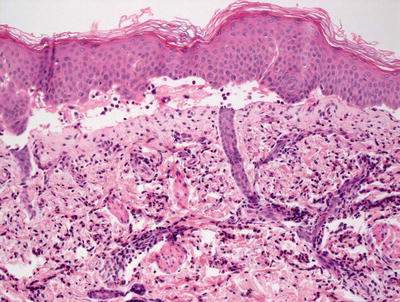
Fig. 3.33
Recessive dystrophic epidermolysis bullosa demonstrates a subepidermal blister with some lymphocytic inflammation
3.13.3 Pathogenesis
Dystrophic epidermolysis bullosa is caused by mutations in COL7A1 gene, which encodes collagen VII [123]. To date, several hundred mutations in COL7A1 have identified in both recessive and dominant forms of the disease [124, 125]. Mutational analysis of COL7A1 gene can be performed by PCR amplification of all 118 exons and exon–intron boundaries as well as by direct DNA sequencing [126]. The blisters in dystrophic epidermolysis bullosa are caused by structural and functional abnormalities in the anchoring fibrils that serve to anchor the epidermal basement membrane to the dermis. Type VII collagen is a major component of anchoring fibrils [112]. Reduction or absence of collagen VII results in the loss of anchoring fibrils and blister formation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


