Chapter 15 Vasculitis
1. How are vasculitic disorders defined and classified?
Vasculitis is defined as inflammation of blood vessels. Vasculitis may be confined to the skin; however, the majority of cases of cutaneous vasculitis are part of multisystemic disorders that in addition to involving skin also involve other organ systems. Classification is problematic due to the lack of standardization and definition. The most accepted classification scheme for systemic vasculitis syndromes is based on the size of the involved blood vessels, as shown in Table 15-1. Subclassification of these syndromes is based on clinical and histologic criteria that have been determined to be suggestive of a specific disorder. The American College of Rheumatology Subcommittee on Classification of Vasculitis has determined classification criteria for many of these disorders (see Table 15-1). This classification system is excellent for systemic vasculitis but it omits some forms of vasculitis that are confined to the skin.
2. Are there specific serologic markers for any of these vasculitic disorders?
Yes. Antimyeloperoxidase antibodies directed against cytoplasmic components of neutrophils have been used to help identify patients with segmental necrotizing glomerulonephritis and some types of systemic vasculitis. Antibodies directed against serine proteinase 3 that is found in the cytoplasm of neutrophils (c-ANCA) have been detected in 66% to 90% of patients with active Wegener’s granulomatosis. Patients with pulmonary-renal syndrome who have antibodies directed against cytoplasmic myeloperoxidase, in neutrophils that produce a peripheral antineutrophil cytoplasmic pattern (p-ANCA), are most likely to have microscopic polyangiitis.
Table 15-1. Classification of Systemic Vasculitides
| VESSEL SIZE | VASCULITIC SYNDROME |
|---|---|
| Large vessel vasculitis | Giant cell (temporal) arteritis Takayasu arteritis |
| Medium vessel vasculitis | Polyarteritis nodosa (classic PAN) Kawasaki disease |
| Small vessel vasculitis | Wegener’s granulomatosis Churg-Strauss syndrome Microscopic polyangiitis (polyarteritis) Henoch-Schönlein purpura Essential cryoglobulinemic vasculitis Cutaneous leukocytoclastic vasculitis |
Adapted from Jennette JC, Falk RJ, Andrassy K, et al: Nomenclature of systemic vasculitides: proposal of an International Consensus Conference, Arthritis Rheum 37:187–192, 1994. Adopted by the Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis, 1994.
3. What is a leukocytoclastic vasculitis?
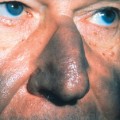
Patients with leukocytoclastic vasculitis, also referred to as leukocytoclastic angiitis and allergic or necrotizing vasculitis, present with characteristic purpuric papules, most frequently involving the extremities, known as palpable purpura (Fig. 15-1). Biopsies of cutaneous leukocytoclastic vasculitis demonstrate an intense perivascular infiltrate composed of intact and fragmented neutrophils (nuclear dust) that focally infiltrate the vessel wall producing fibrinoid changes and/or necrosis. These damaged vessels frequently demonstrate extravasation of erythrocytes and may also demonstrate thrombosis.
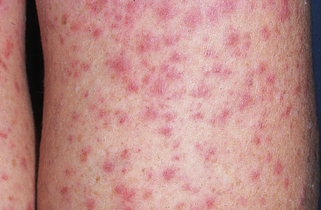
Figure 15-1. Leukoclastic vasculitis secondary to ampicillin. Typical lesions of palpable purpura are seen.
4. What are some important precipitating causes of small vessel leukocytoclastic vasculitis?
• Infections: Bacterial (streptococcal infections, bacterial endocarditis), viral (parvovirus B19, HIV, hepatitis A–C), mycobacterial (Hansen’s disease, tuberculosis), fungal (Candida albicans), protozoan (Plasmodium malariae), helminthic (Schistosoma haematobium, S. mansoni, Onchocerca volvulus)
5. What is Henoch-Schönlein purpura?
Henoch-Schönlein purpura is a variant of leukocytoclastic vasculitis characterized by the deposition of immune complexes containing IgA in small vessels with the most commonly affected vessels being in the skin, kidney, and gastrointestinal tract. The precipitating antigen is not identifiable in all cases but many cases are associated with streptococcal or viral infections. It is usually seen in young children although any age can be affected. The classic four components of the syndrome include cutaneous palpable purpura, colicky abdominal pain, arthralgias and/or arthritis and kidney involvement.
6. What is the mnemonic that can help remember the clinical features of Henoch-Schönlein purpura?
The mnemonic PAPAH (pä-pä) can be used to remember the clinical features of HSP: purpura, abdominal pain, arthralgias, hematuria. Some patients with abdominal involvement demonstrate significant melena or less commonly develop intussusception of the bowel. While more than 40% of patients demonstrate at least some degree of renal involvement, primarily manifesting as hematuria and proteinuria, only 1% develop chronic renal impairment. Less commonly, other organs such as the brain or lungs can be involved.
7. What is “acute hemorrhagic edema of infancy” and how does it differ from Henoch-Schönlein purpura?
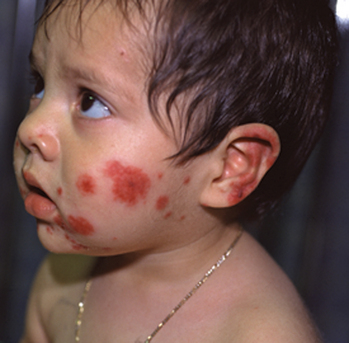
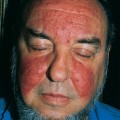
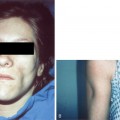
Acute hemorrhagic edema of infancy, which also called “cockade purpura,” is also an IgA-mediated immune complex leukocytoclastic vasculitis that affects small cutaneous vessels. This variant almost always affects young children (median age is 11 months), and like Henoch-Schönlein purpura, it is usually associated with a preexisting upper respiratory infection. It differs in that the primary cutaneous clinical lesions are typically indurated, edematous plaques with variable hemorrhage that are less likely to ulcerate. The lesions frequently affect the head and neck region and extremities (Fig. 15-2). The children typically do not demonstrate significant involvement of the gastrointestinal tract, joints or kidneys and do not develop permanent sequelae.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


