, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
21.1 Capillary Malformations
21.1.1 Cutaneous and Mucosal Capillary Malformations (Port-Wine Stains)
21.1.1.1 Clinical Features
Capillary malformations (also known as port-wine stains) are superficial vascular malformations. They occur in 0.3–0.5 % of the population [1]. Capillary malformations appear as pink to erythematous or violaceous macules and patches (Fig. 21.1). They can occur anywhere on the body, but are most common on the neck and head, particularly the face. They may darken and thicken over time. Port-wine stains can occur in isolation or as part of a syndrome. Syndromic presentations include Sturge–Weber syndrome with central nervous system lesions, Klippel–Trenaunay–Weber syndrome with limb hypertrophy, and phakomatosis pigmentovascularis with a variety of melanocytic lesions [2–6]. The melanocytic lesions can include nevus of Ota, nevus spilus, congenital dermal melanocytosis, and melanosis oculi. Capillary malformations can be treated with a variety of vascular-specific lasers, particularly pulsed-dye laser.
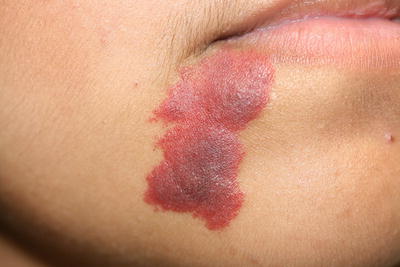
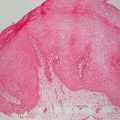
Fig. 21.1
Capillary malformation may appear as a thin, well-defined red patch or plaque on the face
21.1.1.2 Histology
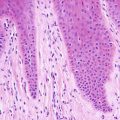
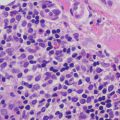
Capillary malformations have variable histologic patterns, depending upon the age of the lesion. Early lesions demonstrate only subtle histologic changes, and could be misinterpreted as normal skin. There is minimal vascular dilatation of small vessels in the papillary and superficial reticular dermis. Vascular congestion is prominent. With increasing age, the vessels progressively dilate, initially remaining thin-walled and demonstrating a single layer of flattened endothelial cells (Fig. 21.2). Eventually, the vascular walls have increased thickening from compensatory hyperplasia due to increased blood flow. The vessel walls can attain marked thickening resulting in a nodular appearance [7, 8].
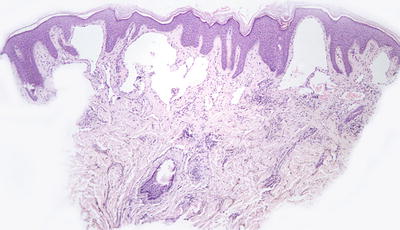
Fig. 21.2
Dilated capillaries are present in the superficial dermis in capillary malformations. They may be congested in older lesions
The incidence of misdiagnosis of pediatric vascular neoplasms is high, and attention to the differential diagnostic features is crucial [9]. The histologic differential diagnosis includes infantile hemangioma. Capillary malformations fail to demonstrate the focality or lobularity seen in an infantile hemangioma, and they are diffuse across the entire biopsy specimen. Later lesions may be confused with arteriovenous malformations due to the thickened vessels walls. Arteriovenous malformations tend to have increased vascular density, more vascular luminal red blood cells, more branching of dermal vessels, and thicker vessels than are seen in capillary malformations [10]. Some acquired port-wine stains can appear post-traumatically, providing evidence to the hypothesis that these vascular malformations are the result of focal defects in autonomic innervation [11–15]. An eczematous inflammatory reaction pattern has been described in association with capillary malformations [16]. Angiolymphoid hyperplasia with eosinophilia has developed within the vascular ectasia of a port-wine stain [17].
21.1.1.3 Pathogenesis
Port-wine stain is a superficial cutaneous vascular malformation of capillaries. Abnormal alterations in the sympathetic vascular innervation of the capillaries in capillary malformations have been postulated to play a role in the development of these vascular lesions [15].
Significant insights into the pathogenesis of capillary malformations/port-wine stains came with the finding of an underlying somatic mutation in non-syndromic port-wine stains and in those associated with Sturge–Weber syndrome. The mutation is a single-nucleotide variant (c.548G > A, p.Arg183Gln) in the GNAQ gene, in which 88 % of patients with Sturge–Weber syndrome and 92 % of patients with non-syndromic port-wine stains harbor the mutation [18]. Guanine nucleotide-binding protein (G protein) Q polypeptide [GNAQ] gene encodes an α-subunit in the Gq class [19]. GNAQ is involved in cell signaling to activate mitogen-activated protein kinase (MAPK) and phospholipase C pathways to regulate cell proliferation and functions. GNAQ p.Arg183Gln substitution can activate ERK/MAPK signaling pathway, a mechanism that can lead to the capillary malformations seen in port-wine stains and Sturge–Weber syndrome [18]. In addition to the known mutation in GNAQ, somatic mutations in a number of other genes, namely, SMARCA4, EPHA3, MYB, PDGFR-β, and PIK3CA, have been found in a single case of isolated port-wine stains [20].
Inactivating mutations in Ras p21 protein activator 1 (RASA1) gene have been identified in some families with multiple capillary malformations , including those with capillary malformation–arteriovenous malformation syndrome (CM–AVM) [21–23]. RASA1 encodes for p120 Ras-GAP, which is a GTPase-activating protein that negatively regulates Ras activity. Loss-of-function mutations of RASA1 lead to increased Ras activation and unregulated cell proliferation, including vascular growth.
Pulsed dye laser (PDL) is a gold standard of treatment for capillary malformations. PDL induces an increase in hypoxia-inducible factor (HIF)-1α, vascular endothelial growth factor, and activated ribosomal protein S6 (pS6), indicating that angiogenesis pathways are involved in skin blood vessel regeneration and revascularization [24]. These angiogenic effects can be blocked by rapamycin (sirolimus), which is an inhibitor of the mammalian target of rapamycin (mTOR) , implicating a role of the mTOR pathway in port-wine stains [25]. Studies of combined therapy of PDL and rapamycin to inhibit neovascularization in port-wine stains following laser ablation have shown that the combination approach provides a novel way to block lesion recurrence [26–28]. A phase II clinical trial of combination therapy of topical rapamycin and PDL has shown this to be an effective treatment for port-wine stains [29].
21.1.2 Sturge–Weber Syndrome
21.1.2.1 Clinical Features
Sturge–Weber syndrome (SWS) (also known as encephalofacial angiomatosis) is a sporadic congenital condition characterized by facial capillary malformations (port-wine stains) in the V1 distribution of the trigeminal nerve, ipsilateral glaucoma, and venous-capillary abnormalities of the leptomeninges (Fig. 21.3) [30]. A child born with port-wine stains located in the distribution of the V1 branch of the trigeminal nerve has a 26 % risk of having Sturge–Weber syndrome [31]. Patients often have seizures and other neurologic problems.
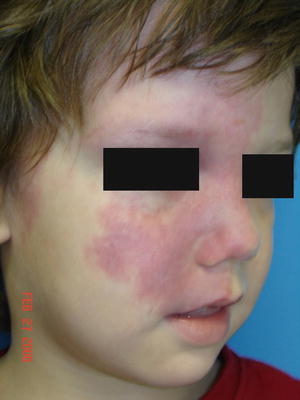
Fig. 21.3
Sturge–Weber syndrome patient with a large pink patch (port-wine stain) primarily involving the V1 distribution of the face. The child also has seizures and glaucoma (photo courtesy of Marcelo Hochman, MD, Hemangioma & Malformation Treatment Center, Charleston, SC)
21.1.2.2 Histology
The histologic changes of port-wine stains that occur sporadically and as part of Sturge–Weber syndrome are identical. Some patients with Sturge–Weber syndrome also demonstrate pigmented lesions and can be classified as having phakomatosis pigmentovascularis, type IIb [2].
21.1.2.3 Pathogenesis
An underlying somatic mutation has been identified in Sturge–Weber syndrome. The mutation is a single-nucleotide variant (c.548G > A, p.Arg183Gln) in GNAQ gene with 88 % of patients with Sturge–Weber syndrome harboring the mutation [18]. GNAQ [guanine nucleotide-binding protein (G protein), Q polypeptide] gene codes an α-subunit in the Gq class [19]. GNAQ is involved in cell signaling to activate mitogen-activated protein kinase (MAPK) and phospholipase C pathways to regulate cell proliferation and vascular growth . GNAQ p.Arg183Gln substitution causes increased activation of the ERK/MAPK signaling pathway, a mechanism that can lead to the capillary malformations and tissue overgrowth seen in Sturge–Weber syndrome [18].
21.2 Venous Malformations
21.2.1 Common Venous Malformations
21.2.1.1 Clinical Features
Venous malformation is a congenital vascular anomaly that is present at birth and continues to grow with the child. Venous malformation consists of collections of abnormal veins. It can be localized or diffuse, and often presents as a soft, compressible bluish mass [32]. Due to low blood flow, venous malformation is prone to the development of intravascular thrombi, which may calcify to form phleboliths. Localized intravascular coagulopathy with elevated D-dimers is a characteristic of the lesion [33, 34]. Venous malformation grows and enlarges over time, and can cause substantial morbidity because of disfigurement, bleeding, and pain. Sclerotherapy is a first-line treatment of venous malformation, although it is not entirely effective [32, 35]. Therapy is typically aimed at alleviation of symptoms as many lesions are too extensive for complete surgical resection.
21.2.1.2 Histology
Venous malformation is composed of enlarged blood vessels lined by flat endothelial cells with irregular lumen size and shape. The lesion often involves the dermis and subcutaneous adipose tissue. The vascular wall has a variable thickness of smooth muscles, and some areas lack smooth muscle altogether [36, 37]. Intravascular thrombi and phleboliths are common. Areas of fibrosis, red blood cell extravasation, and hemosiderin deposition are frequent findings .
21.2.1.3 Pathogenesis
Sporadic venous malformation is caused by somatic mutation in TEK2 gene, which encodes endothelial cell receptor tyrosine kinase TIE2 [38–40]. TIE2 mutations are found in about half of the cases of venous malformations. The most common TIE2 hot-spot mutation in sporadic venous malformations is the somatic mutation L914F, which accounts for over 70 % of patients with TIE2 mutations [38, 39, 41]. Hereditary venous malformation (as in familial mucocutaneous venous malformation) is also caused by TIE2 mutations, and the most common inherited mutation is R849W [38, 40, 42]. These mutations cause constitutive activation of TIE2, leading to chronic activation of Akt signaling pathway, and abnormal cell proliferation and differentiation. TIE2 signaling is also critical for communications between endothelial cells and smooth muscle cells in venous morphogenesis [40].
The importance of TIE2 in the development of venous malformation has been shown in experimental animal models. Normal human endothelial cells expressing the TIE2 mutation L914F develop lesions similar to human venous malformation in a mouse model [43]. Rapamycin (sirolimus), which is a selective inhibitor of mammalian target of rapamycin (mTOR) , prevents venous malformation growth in this model. Extending these findings into a prospective clinical pilot study, rapamycin has been shown to reduce pain, bleeding, and functional impairment in six patients with venous anomalies [43]. Inhibition of the Akt/mTOR signaling pathway with rapamycin provides some clinical improvement in patients with venous malformation.
Not all venous malformations carry TIE2 mutations. Somatic mutations in the PIK3CA gene have been found in over 50 % of venous malformations with no detectable TIE2 mutations [44–46]. The hot-spot mutations are c.1624G > A, c.1633G > A, and c.3140A > G (p.Glu542Lys, p.Glu545Lys, and p.His1047Arg), and they account for more than 90 % of individuals with venous malformations who carry PIK3CA mutations. PIK3CA encodes for the p110-α catalytic subunit of PI3-Kinase [47]. PIK3CA mutations cause constitutive activation of the Akt signaling pathway, leading to dysregulation of important angiogenic factors and abnormal endothelial cell proliferation [46, 47]. A genetically-engineered animal model with mosaic expression of constitutively active PIK3CA (H1047R) mutation recapitulates features of human venous malformation [45]. Rapamycin treatment can induce venous malformation regression in animal models. PIK3CA and TIE2 mutations appear to operate via the same pathogenic pathways leading to Akt activation. However, there are gene-specific effects between PIK3CA and TIE2 mutations, since there is significant genotype–phenotype correlation in lesion localization and histology between these two genes [46].
21.2.2 Blue Rubber Bleb Nevus Syndrome
21.2.2.1 Clinical Features
Blue rubber bleb nevus syndrome (BRBNS) is a rare condition characterized by multifocal cutaneous and visceral venous malformations [48]. Lesions are often present at birth, but may also appear in childhood or adulthood. Some lesions may enlarge over time. Venous malformations in the small intestine can cause serious acute and chronic blood loss. Cutaneous lesions appear as blue papules and compressible nodules (Fig. 21.4). Prognosis depends on the extent of disease and the organs involved.
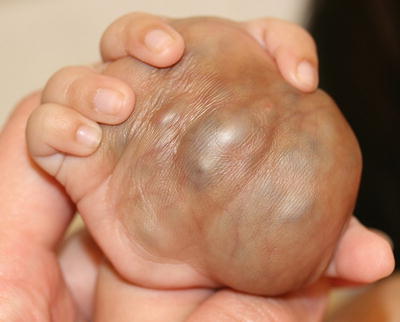
Fig. 21.4
A large venous malformation is seen in the palm of an infant with blue rubber bleb nevus syndrome
21.2.2.2 Histology
Histologic changes in BRBNS are those of venous malformations. While earlier literature suggested these lesions to be cavernous hemangiomas [49], current classifications define the dilated vessels as veins [48]. Large dilated, thin-walled veins are present within the reticular dermis and extending into the subcutis [50]. The blood vessels may be surrounded by fibrosis, and some vessels may have slight thickening in the vascular walls. Occasional vessels may be congested, or if they are traumatized, thrombi and intravascular papillary endothelial hyperplasia (Masson’s lesions) may be present [51]. A rare case of a blue rubber bleb nevus with an infiltrative lymphangiomatosis-like growth pattern and focal podoplanin (lymphatic marker) expression has been reported [52]. Similar venous malformations may be seen in the viscera, including the gastrointestinal tract where bleeding can be clinically significant [50, 53, 54]. Some syndrome cases have been associated with leukemia and visceral tumors [55].
21.2.2.3 Pathogenesis
According to the International Society for the Study of Vascular Anomalies (ISSVA) classification, BRBNS is a disease of multifocal venous malformations primarily involving the skin and gastrointestinal tract [56]. Most cases are sporadic. However, several familial cases have been identified, and have an autosomal pattern of inheritance [57, 58].
Activating mutations in TIE2, a receptor tyrosine kinase highly expressed in endothelial cells, have been identified in some cases of BRBNS [40, 59]. BRBNS also has been found to harbor mutations in c-Kit (stem cell growth factor receptor), which is a tyrosine kinase that activates mTOR (mammalian target of rapamycin) [60]. mTOR is a key mediator of growth factor receptor signaling, including the proangiogenic vascular endothelial growth factor [61]. BRBNS is highly responsive to treatment with rapamycin (sirolimus ), which is a potent and selective inhibitor of mTOR [62, 63]. Even though BRBNS is primarily a venous malformation, it also has a lymphatic component and shows cell proliferative activity based on staining for MIB-1. Together with the findings of increased receptor tyrosine kinase signaling and activation of mTOR signaling, this may be a possible explanation for the therapeutic response seen with rapamycin in BRBNS, since rapamycin has been consistently demonstrated as being effective in the management of complicated vascular malformations with lymphatic component [62].
21.3 Lymphatic Malformations
21.3.1 Microcystic Lymphatic Malformations (Lymphangioma Circumscriptum)
21.3.1.1 Clinical Features
Microcystic lymphatic malformations (lymphangioma circumscriptum) are a superficial variant of cutaneous lymphatic malformation. Most lesions typically are noticed at or soon after birth [64]. Common locations include axillary folds, shoulders, proximal extremities, and genitalia. Cutaneous lesions appear as clusters of vesicles containing clear, yellow, pink, or hemorrhagic fluid (Fig. 21.5). They are often asymptomatic, but can exude clear or bloody fluid, especially after trauma. Areas may become infected. Treatment modalities include surgical excision and other destructive therapies, such as laser ablation, cryotherapy, and sclerotherapy. Recurrence is common.
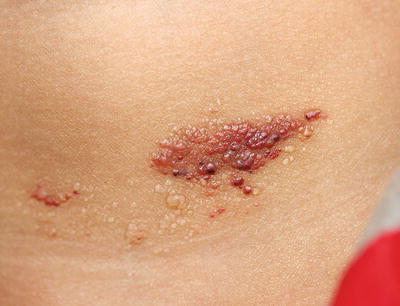
Fig. 21.5
Lymphangioma circumscriptum presents as a cluster of small clear and hemorrhagic fluid-filled vesicles
21.3.1.2 Histology
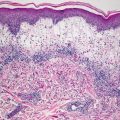
Histologic features of microcystic lymphatic malformation include an epidermis that is focally atrophic with elongated rete ridges that extend down and surround superficially located and dilated vascular channels [65–67]. The epidermis directly overlying the vascular channels is often atrophic, but rare cases of verrucous epidermal hyperplasia with papillomatosis and acanthosis have been described [66, 68]. The ectatic vessels are thin-walled and lined by flattened endothelial cells (Figs. 21.6 and 21.7). The spaces are often filled with eosinophilic proteinaceous material. When traumatized, hemorrhage into the vascular spaces may be present. Lymphocytes surround the dilated vessels in some cases. Dilated lymphatic spaces may extend into the deeper dermis and subcutaneous fat, but this feature is not seen on all biopsies [65, 67, 69, 70]. Ultrastructural analysis has suggested that the primary pathology of these lesions resides in the deep and enlarged malformed vessels, and the superficial vessels represent secondary changes of long-standing lymphostasis [71].
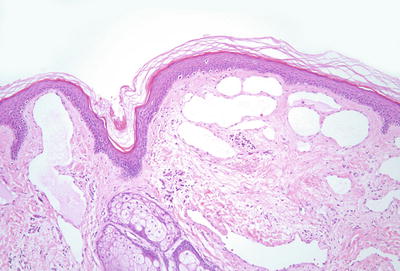
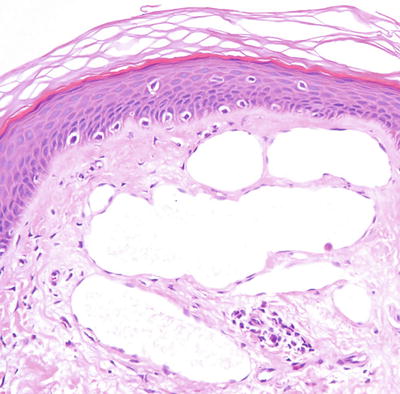
Fig. 21.6
Within the superficial dermis, large thin-walled, and dilated vascular channels are surrounded by acanthotic epidermis in lymphangioma circumscriptum
Fig. 21.7
Vessels are lined by flattened endothelial cells and often do not contain erythrocytes in lymphangioma circumscriptum
A single case study of Dabska tumor arising within a lymphangioma circumscriptum has been reported [72]. The relationship between lymphangioma circumscriptum and lymphangioendothelioma, which is a more cellular and invasive lesion, is still uncertain, since they both appear to be lymphatic malformations and not neoplastic processes [73].
The main histologic differential diagnosis is angiokeratoma, especially when trauma results in hemorrhage into the vascular spaces. Clinical correlation often helps in resolving the diagnostic dilemma. D2-40 immunostain that labels lymphatic endothelial cells can also be used in most cases to establish a definitive diagnosis. A newly identified lymphatic malformation has been described that is characterized by dilated and thin-walled vascular channels in the upper dermis. Prominent endothelial cells protrude into the lumen. In this lesion, there is no epidermal change identified, and the clinical presentation is distinct from lymphangioma circumscriptum. Based upon the clinical appearance, the authors have suggested the name lymphatic netlike malformation [74].
21.3.1.3 Pathogenesis
It has been proposed that microcystic lymphatic malformation may be caused by abnormal lymphatic cisterns that grow separately from normal lymph vessels during development [66, 67]. Contractions of the smooth muscles lining the cisterns can cause the formation of abnormal and dilated lymphatic vessels.
Molecular studies of a patient with a lymphatic malformation show that endothelial cells within the lymphatic malformation harbor mutations in PI3-Kinase with associated hyperactivation of downstream Akt signaling pathway [75, 76]. Moreover, treatment with rapamycin (sirolimus ), which is an inhibitor of the mammalian target of rapamycin (mTOR) , blocks the proliferation and sprouting angiogenesis of lymphatic malformation cells, implicating a role for PI3-Kinase/Akt/mTOR signaling in lymphatic malformations .
21.3.2 Macrocystic Lymphatic Malformations (Cavernous Lymphangioma)
21.3.2.1 Clinical Features
Macrocystic lymphatic malformation (alternative names include cavernous lymphangioma, cystic lymphangioma, and cystic hygroma) is typically seen on the head, neck, or axilla of newborn infants [77]. An unusual variant may affect the abdominal viscera and cavity [78]. Most lesions present as a large soft mass in the neck or axilla. Problematic lesions are managed with sclerotherapy and surgical excision .
21.3.2.2 Histology
In macrocystic lymphatic malformation, deep-seated dilated vessels are present in the deep dermis and often extend into the subcutaneous fat [65]. The vessels are lined by flattened endothelial cells [79]. However, in some cases, underlying lymphoid nodules result in endothelial cell protrusions into the vascular lumen. Increased fibrosis may result in vascular wall thickening in some lesions. Vascular valves are observed in some cases, and due to trauma, hemorrhage may be encountered within vessels and in the surrounding dermis. When extensive hemorrhage is present, differentiation from other types of vascular malformations can be difficult. D2-40 immunostain, which is a lymphatic marker, is sometimes helpful in resolving this diagnostic dilemma based upon the strong staining of lymphatic endothelial cells.
The relationship between macrocystic lymphatic malformation and microcystic lymphatic malformation is not fully understood. Microcystic lesion demonstrates widely ectatic lymphatic vessels in the superficial dermis. However, in some cases, there is extension of the malformation into the deep dermis, subcutis, and even musculature. In these cases, the deeper components are indistinguishable from those seen in macrocystic lesion, thus blurring the histologic distinction between microcystic and macrocystic malformations [66, 70].
21.3.2.3 Pathogenesis
Microcystic and macrocystic lymphatic malformations share similar pathogenesis (see previous section of the pathogenesis for microcystic lymphatic malformation ).
21.3.3 Primary Congenital Lymphedema (Milroy Disease)
21.3.3.1 Clinical Features
Primary congenital lymphedema (Milroy disease) is a disorder with an autosomal dominant inheritance, and disease incidence is approximately 1:6000 births [80]. Infants present prenatally or perinatally with tense edema of one or both feet and legs. Swelling is typically painless and non-pitting. Complications are rare, but bacterial infections and discomfort are possible. Long-term complications of massive congenital lymphedema include development of angiosarcoma [81]. There is no definitive treatment. However, compression garments and lymphatic massage may be helpful.
21.3.3.2 Histology
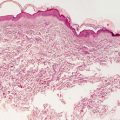
Histologic changes are primarily in the dermis and subcutis. Overlying epidermal changes are secondary, and may include verrucous epidermal hyperplasia, epidermal acanthosis and papillomatosis, and overlying hyperkeratosis. Widened intercellular spaces are present as evidenced by increased spaces between collagen bundles. There is no increase in cellularity, and in most cases, the lymphatic channels are atrophic so that endothelial cells are not readily apparent [82] (Figs. 21.8 and 21.9).
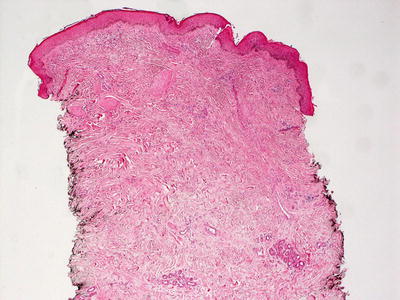
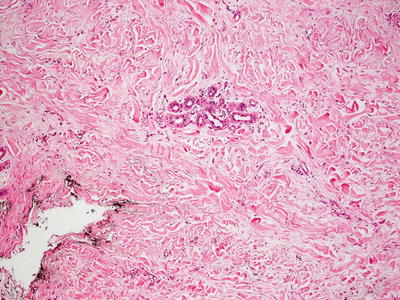
Fig. 21.8
Milroy lymphedema gives rise to a markedly thickened dermis with densely fibrotic stroma and relatively inconspicuous thin-walled vascular channels
Fig. 21.9
Dermal collagen is fibrotic and often demonstrates areas with increased interstitial edema in Milroy lymphedema. It is often difficult to identify the affected vessels
Pigmented purpuric eruption has been described in a patient with Milroy disease. It is unclear whether there is any true association other than coincidental coexistence of these two entities [83]. Verruciform xanthoma has been described with overlying massive lymphedema in some patients. These changes are not unexpected as being secondary to verrucous epidermal hyperplasia seen in some cases of chronic lymphedema [84, 85].
21.3.3.3 Pathogenesis
Primary congenital lymphedema is an autosomal dominant genodermatosis caused by dysfunction of the lymphatic system. A known causative gene is vascular endothelial growth factor receptor-3 (VEGFR-3) [86–89]. Although not all individuals with the disease have identifiable mutations in VEGFR-3, in one case series, 90 % of 71 individuals found to carry a VEGFR-3 mutation had signs of Milroy disease with edema confined to the lower legs [86, 90]. Another study indicated that VEGFR-3 mutations have been found in 75 % of patients with familial Milroy disease, and in 68 % of patients with sporadic disease [91]. Over 50 different mutational variants in VEGFR-3 have been observed [92]. Most of the mutations are missense mutations with a few deletions and splicing variants.
VEGFR-3 is an important gene in the development and growth of lymphatic vessels. It is a receptor tyrosine kinase, and is activated when bound to vascular endothelial growth factor-C (VEGF-C) and VEGF-D [93, 94]. Mutations in the tyrosine kinase domains of VEGFR-3 are sufficient to reduce tyrosine kinase activity and affect lymphatic development [89]. The importance of VEGFR-3 in lymphedema has been demonstrated in an animal model, in which transgenic mice with inhibition of VEGFR-3 develop hind limb lymphedema similar to that seen in Milroy disease [95].
21.4 Arteriovenous Malformations
21.4.1 Sporadic Arteriovenous Malformations
21.4.1.1 Clinical Features
Arteriovenous malformations (AVM) are congenital vascular malformations that contain variable degrees of arteriovenous shunting. They have a tendency to worsen and become more problematic over time. Focal lesions may expand rapidly and ulcerate. They can also result in cardiac failure, hemorrhage, and stroke. The clinical appearance of AVM varies depending on the size and extent of the lesion [96, 97]. Some present as a pink patch that resembles a capillary malformation, while others may appear more blue and nodular. AVM typically feels warm to the touch, and has a palpable thrill with audible bruit when examined (Fig. 21.10).
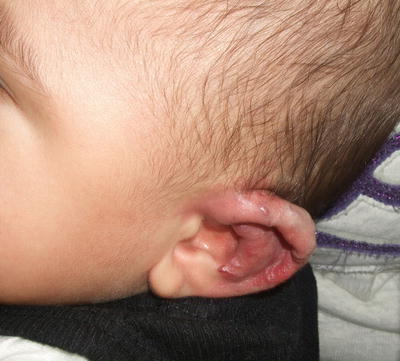
Fig. 21.10
A violaceous nodular plaque is seen on the ear of an infant with an arteriovenous malformation
21.4.1.2 Histology
Histologic findings vary with the types of vessels involved in arteriovenous malformation. Combinations of lymphatic, venous, capillary, arterial, and glomus cells have all been described in malformations in the skin [98–101]. Thin-walled vessels can be seen admixed with thicker-walled vessels surrounded by smooth muscle (Figs. 21.11 and 21.12). In other cases, thin-walled vessels are lined by endothelial cells that label focally as venous, and in other regions as lymphatic vascular channels [102]. In general, these lesions tend to have widely dilated vascular channels at least in some foci, and tend to extend through the dermis into the subcutis.
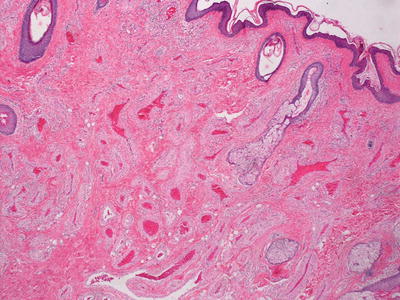
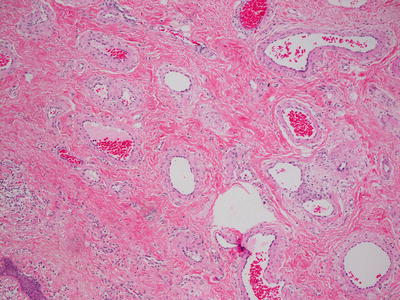
Fig. 21.11
AVM demonstrates increased numbers of dermal blood vessels, some of which have thin walls and others have thickened walls, resembling arterioles
Fig. 21.12
A collection of dermal blood vessels with vascular walls of varying thickness in an AVM
The histologic differential diagnosis includes vascular proliferative processes, and it is dependent upon the various components present within the malformation. A rare variant of capillary arteriovenous malformation with ancient or symplastic change demonstrates cytologic atypia, and it could be mistaken for a malignant vascular neoplasm [103].
21.4.1.3 Pathogenesis
Arteriovenous malformation can occur sporadically or in association with inherited disorders, such as hereditary hemorrhagic telangiectasia (HHT) and capillary malformation–arteriovenous malformation syndrome (CM–AVM) [97]. HHT is an inherited vascular disorder with AVM involving the skin and mucosa, and has significant morbidity [104, 105]. The genetic cause of HHT is mutations in endoglin (ENG), activin A receptor type II-like 1 (ACVRL1), and SMAD4 [106–108]. Importantly, these genes are all involved in various aspects of transforming growth factor-β (TGF-β) signaling pathway.
CM–AVM is a vascular malformation syndrome with capillary malformations of the skin, and they are often associated with underlying AVM [109]. CM–AVM is caused by mutations in RASA1 (Ras p21 protein activator 1) gene [21, 22]. RASA1 encodes for p120 Ras-GAP, which is a GTPase-activating protein that negatively regulates Ras activity [110]. Loss-of-function mutations of RASA1 lead to increased Ras activation and unregulated cell proliferation, including vascular growth .
21.4.2 Cobb Syndrome
21.4.2.1 Clinical Features
Cobb syndrome is characterized by the presence of a spinal arteriovenous malformation and an overlying cutaneous vascular anomaly in the same dermatome [111]. Cobb syndrome is quite rare, but usually presents in late childhood with neurological symptoms [112]. Disease management varies but may include surgery and endovascular embolization.
21.4.2.2 Histology
21.5 Vascular Malformations Associated with Other Anomalies
21.5.1 Klippel–Trenaunay Syndrome
21.5.1.1 Clinical Features
Klippel–Trenaunay syndrome (KTS) is a congenital vascular disorder with capillary malformations, venous malformations, and hypertrophy of the affected tissues [119, 120]. Capillary malformations (port-wine stains) are the most common cutaneous vascular anomaly seen in KTS. Port-wine stains are red to purplish in color, and can be present on any part of the body. Varicose veins and lymphatic malformations with subsequent nodular hypertrophy of the affected area also occur in patients with KTS. Vascular malformations in visceral organs can cause severe bleeding. Limb hypertrophy, mostly in the leg, occurs in the majority of patients [121] (Fig. 21.13).
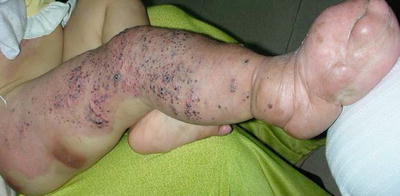
Fig. 21.13
Klippel–Trenaunay syndrome presents with characteristic soft tissue overgrowth, capillary malformation, and venous varicosity in the limb
21.5.1.2 Histology
The cutaneous histologic changes of KTS are indistinguishable from those seen in port-wine stains not associated with limb hypertrophy. Ectatic thin-walled capillary vessels are present initially in the superficial dermis. With time, the vascular lesions become progressively dilated, and the vessel walls have greater thickness. Vessels deeper in the dermis are also affected. Thrombi may be present in congested vessels. These lesions can become nodular and are likely to recur [122]. In rare cases, angiokeratomas have been described in patients with KTS [123].
Secondary changes, such as spongiotic dermatitis with inflammation (Meyerson’s phenomenon ) and acro-angiodermatitis (marked stasis changes or pseudo-Kaposi’s sarcoma) are found in some patients with KTS. The spongiotic changes include intraepidermal edema and lymphohistiocytic infiltrate [16]. Acro-angiodermatitis is characterized by proliferation of slit-like vascular spaces and lobular proliferations of markedly thick-walled vessels. There is abundant extravasation of erythrocytes and hemosiderin deposition. The histologic changes are those of prominent stasis dermatitis [124]. A subset of patients with KTS vascular lesions have associated pigmented lesions, and thus have the designation of phakomatosis pigmentovascularis type IIb [2, 125].
21.5.1.3 Pathogenesis
Klippel–Trenaunay syndrome occurs sporadically without a definite mode of familial inheritance. A novel angiogenic gene VG5Q (vascular or vasculogenesis gene on 5q, also called AGGF1 ) was identified as a susceptibility gene of KTS through the presence of activating mutation E133K in some of the KTS patients investigated [126]. However, this finding has since been disputed because the same E133K mutation has been found in healthy individuals as well, suggesting that this is a polymorphism and not a causative mutation [127]. A study of patients with lymphatic malformations and KTS revealed somatic mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) [76]. The study also demonstrated that there is low-level somatic mosaicism in these lesions because of a low abundance of mutant cells in lesional tissue .
21.5.2 Maffucci Syndrome
21.5.2.1 Clinical Features
Maffucci syndrome is a rare condition characterized by multiple venous malformations of the skin and gastrointestinal tract as well as enchondromas, which are benign cartilaginous tumors [128]. Features of the condition typically appear in children by 4 to 5 years of age [128–131]. Skin vascular lesions (usually spindle cell hemangioma) often protrude as soft blue subcutaneous nodules on the distal extremities. These vascular tumors often develop intravascular thrombi and form phleboliths [128, 130, 131]. Other types of vascular lesions, such as venous-lymphatic malformation and hemangioendothelioma, can occur but they are less common. Enchondromas in Maffuci syndrome appear in the phalanges and long bones, and can cause secondary fractures. There is a 30 % risk of an enchondroma undergoing malignant transformation into chondrosarcoma in these patients. Therefore, attention should be paid to lesions that rapidly increase in size with localized swelling and persistent pain, especially at night [129].
21.5.2.2 Histology
Vascular lesions seen in Maffucci syndrome are varied. In addition to venous malformations, lymphatic malformations are also commonly seen [132]. Spindle cell hemangioendothelioma (now reclassified as spindle cell hemangioma) was reported in a patient with Maffucci syndrome [133]. Enchondromas, which are benign cartilaginous tumors comprising of lobules of hyaline cartilage encased by bone and covered by fibrous tissue, are the other classic finding in this syndrome. Several neoplastic processes, including malignant transformation of enchondromas, are seen in patients with Maffucci syndrome. Cutaneous angiosarcoma is reported in a single patient with the syndrome [134].
21.5.2.3 Pathogenesis
Maffucci syndrome is a non-hereditary mesodermal dysplasia. The syndrome carries somatic mutations in IDH1 (isocitrate dehydrogenase 1) and IDH2 (isocitrate dehydrogenase 2) genes [135–137]. The mutation hotspots are R132C and R132H in IDH1 and R172S in IDH2. It has been reported that 77 % patients with Maffucci syndrome carry IDH1 (98 %) or IDH2 (2 %) mutations in their tumors with evidence for intraneoplastic and somatic mosaicism [137]. IDH1 mutations in cartilage tumors are associated with DNA hypermethylation and downregulation of expression of several genes. IDH1 is a metabolic enzyme that generates α-ketoglutarate and NADPH [138]. Gain-of-function mutations in IDH1 may lead to increased production of D-2-hydroxyglutarate. Hydroxyglutarate can exert aberrant epigenetic effects on gene expression by inhibiting histone and DNA demethylation, thus causing hypermethylation and inhibition of gene expression [139, 140]. IDH1 may also activate hypoxia-inducible factor-1α pathway to increase cell proliferation [141].
21.6 Vascular Tumors
21.6.1 Infantile Hemangioma
21.6.1.1 Clinical Features
Infantile hemangiomas are the most common benign tumor of childhood, affecting 4–5 % of Caucasian infants. These lesions are more common in infants who are Caucasian, female, premature and have a history of low birth weight [142]. Infantile hemangiomas can occur anywhere on the skin, but appear to have a predilection for the head and neck region. Occasionally, they may occur in internal organs, particularly the liver.
Clinically, superficial infantile hemangiomas appear as erythematous papules, nodules, masses, and plaques [143] (Fig. 21.14). Deep infantile hemangiomas appear as subcutaneous bluish masses, often with fine erythematous telangiectasias at the surface. Combined infantile hemangiomas are characterized by a deep bluish swelling with overlying erythematous papules and plaques. Infantile hemangiomas are also described as focal or localized lesions arising from a single focus or as segmental with lesions that appear to cover an entire territory of skin. Infantile hemangiomas exhibit a characteristic pattern of behavior, which includes early rapid growth during a proliferative phase, followed by gradual stabilization and slow involution over a period of months to years.
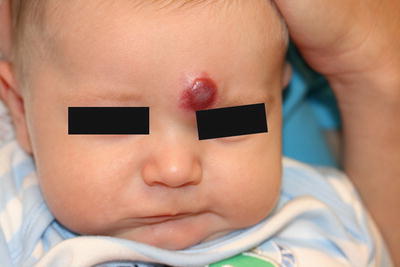
Fig. 21.14
An infantile hemangioma presents as an erythematous nodule on the forehead of an infant
The vast majority of infantile hemangiomas do not require medical intervention. For complex lesions that cause functional impairment or significant cosmetic deformity, oral propranolol is currently the gold standard of medical therapy [144, 145]. Other medications that can sometimes be utilized include systemic corticosteroids, vincristine, and α-interferon. Laser therapy is generally reserved for treatment of ulceration and post-involution residual erythema and telangiectasias . Surgical excision is rarely indicated.
21.6.1.2 Histology
Infantile hemangiomas are densely cellular endothelial cell tumors that occupy the entire dermis, and may extend into the underlying subcutis [146]. Small blood vessels are present with a dense cellularity that includes endothelial cells, pericytes, and stromal fibroblasts [147] (Figs. 21.15, 21.16 and 21.17). Abundant mitoses are present, but there is minimal cytologic atypia. Frequent apoptotic endothelial cells are seen [148]. Many lesions demonstrate extensive areas of dense cellularity with relatively insignificant vascularity; however, on close inspection, areas of vascular proliferation can be detected focally (Fig. 21.18). An infiltrative growth pattern with extension of the hypercellular areas into surrounding collagen and subcutaneous fat causes these lesions to resemble malignant neoplasms [147]. As the lesions progress, the cellularity diminishes, blood vessels become more dilated and lobular in their configuration.
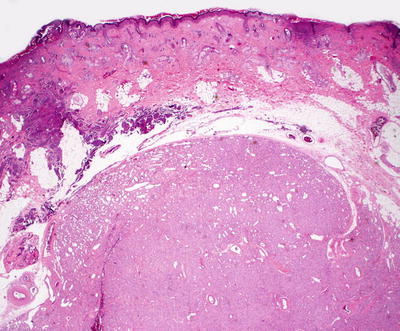
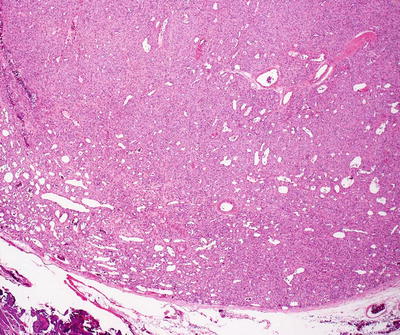
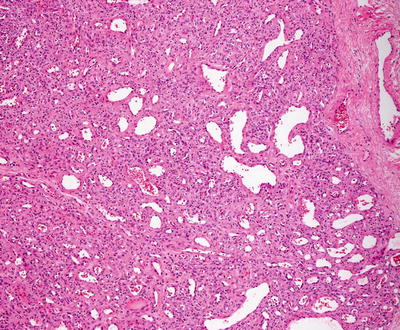
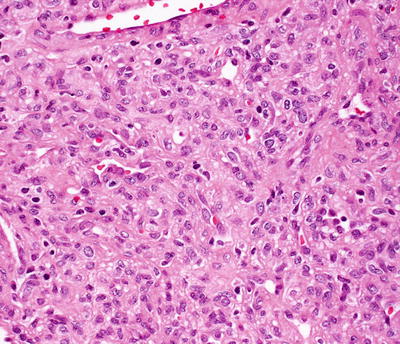
Fig. 21.15
Infantile hemangioma demonstrates densely cellular lobules of small, well-formed blood vessels in the dermis
Fig. 21.16
Well-formed blood vessels and a dense proliferation of ovoid cells are present in infantile hemangioma . The relative amounts of vascularity and surrounding cellularity vary with the age of the lesion
Fig. 21.17
A dense proliferation of blood vessels lined by benign endothelial cells and surrounded by pericytes and stromal cells characterizes the appearance of an infantile hemangioma
Fig. 21.18
A hypercellular proliferation of epithelioid and spindle-shaped cells is present in infantile hemangioma
The differential diagnosis includes spindle cell neoplasms, such as fibrous tumors, neural tumors, and smooth muscle tumors. The identification of vascular differentiation helps to distinguish infantile hemangiomas from other benign and malignant spindle cell tumors. Other cellular vascular neoplasms, such as congenital hemangiomas, may present some diagnostic difficulties. Glucose transporter-1 (Glut-1) immunostaining has been useful in making the distinction, since virtually all infantile hemangiomas strongly and diffusely express Glut-1, whereas congenital hemangiomas are Glut-1 negative (Figs. 21.19 and 21.20) [148]. Pyogenic granuloma may also present a diagnostic challenge, but it can be differentiated based upon the clinical presentation, the architectural changes of a lobular proliferation overlying a single large “feeder vessel,” and the lack of an infiltrative growth pattern. Angiosarcoma and Kaposi’s sarcoma may also be considered, but they demonstrate far more cytologic atypia than that seen in infantile hemangioma. The clinical presentation would exclude these malignant vascular tumors in most cases, but in rare cases where the differential diagnosis includes Kaposi’s sarcoma, HHV-8 immunostains are strongly positive in Kaposi’s sarcoma and negative in infantile hemangioma .
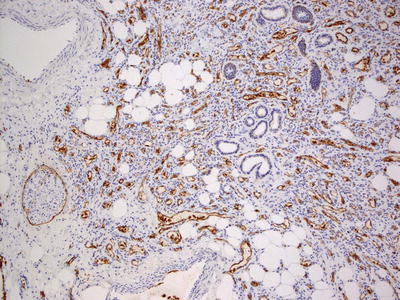
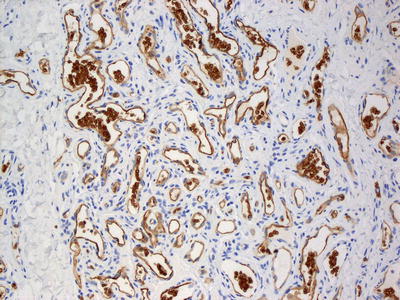
Fig. 21.19
Glut-1 expression is strong and diffuse in infantile hemangioma
Fig. 21.20
Infantile hemangiomas demonstrate strong endothelial cell staining with Glut-1. Note that red blood cells also strongly express Glut-1 on their membranes and serve as a good internal control for staining
21.6.1.3 Pathogenesis
Infantile hemangioma has been well studied in terms of molecular drivers of its development and growth. It has been shown that molecular markers of hemangioma vessels are also expressed in normal placental fetal microvessels, including merosin, laminin, Lewis Y antigen, Fcγ receptor II, and glucose transporter-1 (Glut-1) [149–151]. Glut-1 in particular is strongly expressed in the vessels in infantile hemangioma and placenta, but not in normal skin, vascular malformations, and other types of vascular tumors. Glut-1 serves as a useful diagnostic marker of infantile hemangioma [149]. Global gene expression profiling also shows striking similarities in the mRNA profiles of infantile hemangioma and placenta [152]. The significance of the molecular similarities between infantile hemangioma and placenta is unclear, although it has been postulated that infantile hemangioma may have its origin in the placenta [150–152].
Endothelial cells from proliferating infantile hemangioma appear to be clonal as shown in the patterns of X chromosome inactivation [153, 154]. Multipotent stem cells and endothelial progenitor cells are present in infantile hemangioma. These stem cells can recapitulate features of infantile hemangioma when implanted in an animal model [155, 156].
Somatic mutations in vascular endothelial growth factor (VEGF) receptors have been found in some hemangiomas, and they may lead to abnormal endothelial cell proliferation as a result of abnormal VEGF signaling. A landmark study has shown that infantile hemangioma endothelial cells have low VEGF receptor-1 (VEGFR-1) expression and increased levels of constitutively activated VEGF receptor-2 (VEGFR-2) . VEGFR-1 acts as a decoy receptor that binds to VEGF ligand. The lack of VEGFR-1 decoy receptor results in constitutively activated VEGFR-2 [157]. The downregulation of VEGFR-1 appears to be due to reduced activity of a pathway involving β-1 integrin, integrin-like receptor tumor endothelial marker-8 (TEM8) , VEGFR-2, and nuclear factor of activated T cells (NFAT). Increased VEGFR-2 signaling leads to increased cell proliferation and migration, which can result in the vascular growth in proliferating hemangioma.
VEGF signaling activates a number of proangiogenic signaling pathways, including Akt. Akt has three isoforms (Akt1, Akt2, and Akt3), which have overlapping as well as distinct structures and functions [158]. It has been shown that endothelial Akt1 drives the growth of a variety of vascular tumor cells, including infantile hemangioma endothelial cells [159]. In contrast, Akt3 inhibits vascular tumor growth. Moreover, these opposing effects by different Akt isoforms are mediated by p70 S6-kinase, a protein involved in protein translation. Infantile hemangioma has increased activation of p70 S6-kinase signaling, and loss of the protein reduces hemangioma cell proliferation and migration [160]. p70 S6-kinase is activated by the mammalian target of rapamycin (mTOR) . Inhibition of mTOR with rapamycin (sirolimus) reduces the proliferation of hemangioma endothelial cells as well as the self-renewal potential and differentiation of hemangioma stem cells, leading to the regression of hemangioma blood vessels [160, 161].
Local hypoxia may play a central role in the pathogenesis of infantile hemangioma. There is increased hypoxia-inducible factor-1α (HIF-1α) expression in infantile hemangioma that can lead to increased production of VEGF-A, matrix metalloproteinase-9, and stromal cell-derived factor-1 (SDF-1), all of which can promote hemangioma stem cell trafficking and angiogenesis [162, 163].
Propranolol, which is a nonselective β-adrenergic receptor blocker , has been widely used as the first-line therapy for infantile hemangioma [145, 164, 165]. Despite the well-documented efficacy of propranolol, its mechanism of action has not been fully elucidated. However, the striking efficacy of propranolol points to the importance of the β-adrenergic pathway in infantile hemangioma. The immediate effects of propranolol in hemangioma are likely to be caused by vasoconstriction due to increased contractility of hemangioma pericytes induced by propranolol [166, 167]. These effects manifest as a rapid change in the color and softening of the lesion. The vasoconstrictive effects of propranolol also likely reduce oxygen availability with subsequent tissue hypoxia. The longer-term effects of propranolol may be through blocking receptor-mediated activation of mitogen-activated protein kinase (MAPK), reducing VEGF expression, inhibiting endothelial cell proliferation, and increasing apoptosis [168]. Of note, there is a significant reduction in serum VEGF levels in infants with infantile hemangioma after a few months of propranolol treatment [169, 170]. Propranolol also targets hemangioma stem cells to suppress proliferation and promote apoptosis via β2-adrenergic receptor [171].
Infantile hemangioma undergoes natural regression or involution with replacement of vascular tissue with fibroadipose tissue. A possible mechanism of regression of hemangioma is immune mediated, involving T lymphocytes and indoleamine 2,3-dioxygenase (IDO) [172]. IDO is an enzyme involved in tryptophan metabolism, which is important for the function of T lymphocytes. Low tryptophan levels block T-lymphocyte proliferation and promote apoptosis, thus raising the possibility that inhibition of T lymphocytes through IDO could contribute to the slow regression of hemangioma [173, 174]. Another possible mechanism of hemangioma involution involves mesenchymal stem cells, which are present in infantile hemangioma [175]. These stem cells may differentiate into connective tissue cells and adipocytes to give rise to fibrofatty tissue seen in involuting hemangioma .
21.6.2 Congenital Hemangiomas: Rapidly Involuting Congenital Hemangioma (RICH) and Non-involuting Congenital Hemangioma (NICH)
21.6.2.1 Clinical Features
Congenital hemangiomas are benign vascular tumors that are fully formed at the time of birth. Postnatally, the tumor either involutes quickly (rapidly involuting congenital hemangioma, RICH) or partially regresses and stabilizes (non-involuting congenital hemangioma, NICH) [176, 177]. RICH tends to present as a vascular mass with a surrounding halo of pallor (Fig. 21.21). It typically improves rapidly in the first few months of life. NICH is more likely to present as a telangiectatic patch or plaque, but may also occur as a nodule (Fig. 21.22). NICH does not show a tendency to involute and remains stable over time.
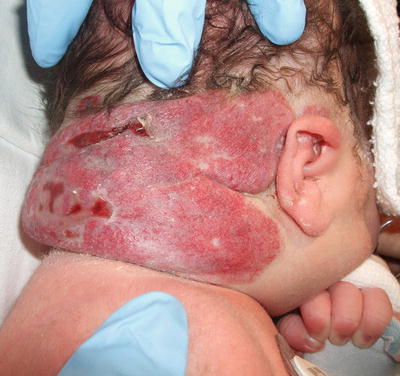
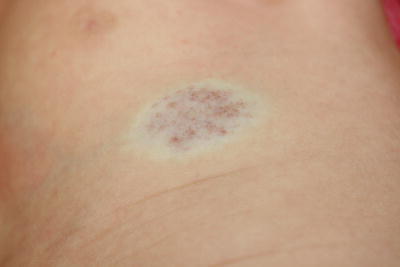
Fig. 21.21
Rapidly involuting congenital hemangioma (RICH) is a large vascular tumor with ulcerations present at birth on the posterior scalp and neck of a newborn
Fig. 21.22
Non-involuting congenital hemangioma (NICH) presents as a thin, telangiectatic plaque with a surrounding blanched “halo”
21.6.2.2 Histology
Both types of congenital hemangiomas can be differentiated histologically from infantile hemangioma.
Rapidly involuting congenital hemangioma is characterized by a lobular proliferation of well-formed blood vessels with surrounding dermal fibrosis. Surrounding pericytes comprise part of the lobular proliferation. Thrombosis is seen within some vessels, and hemosiderin deposition is prominent in some cases. Mitotic activity is relatively scant, and cytologic atypia is not seen in endothelial cells, but they are sometimes characterized as “plump” endothelial cells [176, 178]. Cyst formation, dermal fibrosis , and extramedullary hematopoiesis are seen up to half of cases.
Non-involuting congenital hemangioma is characterized by a lobular capillary proliferation. The lobules are surrounded by dense fibrosis and abundant hemosiderin deposition. The entire lobules may be thrombosed and sclerotic. The neoplastic vessels are sharply circumscribed, and do not appear to be interspersed within the normal dermal vasculature [150, 151]. Arteriovenous fistulas occur within the vascular proliferations, and a large, stellate vessel may appear in the central portion of the lesions [179]. Some authors have described the lobular proliferations as being immediately adjacent to large, malformed vessels [177]. Rarely, these tumors have been associated with multiple epidermal inclusion cysts believed to occur as a consequence of repeated infection [179].
21.6.2.3 Pathogenesis
Genomic DNA and mRNA sequencing of congenital hemangioma tissues revealed somatic activating mutations in the genes GNAQ and GNA11 [181]. Guanine nucleotide-binding proteins (G proteins) are transducers of transmembrane signaling pathways. G proteins are composed of three units: alpha, beta, and gamma. GNAQ encodes for guanine nucleotide-binding protein G(q) alpha subunit, and GNA11 encodes for the guanine nucleotide-binding protein (G protein), alpha-11 subunit [182]. GNAQ shares 90 % protein sequence similarity with GNA11. The mutation found in GNAQ and GNA11 in congenital hemangiomas is a mosaic missense mutation that alters glutamine at amino acid 209 (Gln209) with variant allele frequencies ranging from 3 to 33 %. The same GNAQ or GNA11 mutation is found in both RICH and NICH. The mutation can lead to hyperactivation of downstream pathways, including MAPK, Hippo-Yap, and mTOR pathways [183, 184]. Interestingly, the Gln209 GNAQ and GNA11 missense mutation is also a common mutation in uveal melanoma [185, 186].
Molecular analysis of congenital hemangiomas shows that insulin-like growth factor-2 (IGF-2) is expressed in both RICH and NICH at a level comparable to that detected in infantile hemangioma [187]. In contrast, the expression of vascular endothelial growth factor receptor-1 (VEGFR-1) mRNA is uniformly higher in congenital hemangiomas as compared with infantile hemangioma. These findings indicated molecular similarities as well as distinction between congenital and infantile hemangiomas .
21.6.3 Pyogenic Granuloma (Lobular Capillary Hemangioma)
21.6.3.1 Clinical Features
Pyogenic granuloma, also known as lobular capillary hemangioma, is a common acquired benign vascular lesion. It is most common in adults between 30–40 years of age. In children under age 10, pyogenic granuloma is 2.6 times more common in males than females. Pyogenic granuloma often appears on the head and neck.
Pyogenic granuloma appears as an erythematous papule or mass that is typically pedunculated, and has a tendency to bleed repeatedly and persistently [188] (Fig. 21.23). Since the majority of lesions do not resolve spontaneously, pyogenic granuloma is usually managed with surgical excision or other physical destruction (e.g., cryotherapy, electrocautery, and laser ablation).
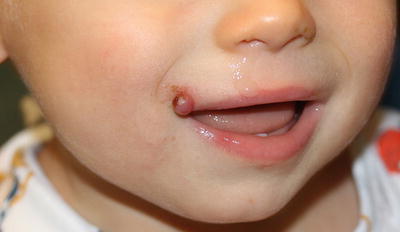
Fig. 21.23
Pyogenic granuloma appears as a pink, pedunculated papule with hemorrhagic crust on the face of a toddler
21.6.3.2 Histology
Pyogenic granuloma has a polypoid architecture with an attenuated or even ulcerated epidermis. There may be an epidermal collarette with elongated rete ridges extending around the lesion in the dermis. A dense lobular proliferation of capillaries is present in the superficial portion of the dermis (Fig. 21.24). In some cases, a larger “feeder” vessel can be identified deep to the lobular proliferation. Small capillaries lined by benign endothelial cells form the tumor with pericytes present around the vessels (Fig. 21.25). Early lesions are densely cellular and may have abundant mitotic activity, but atypical mitoses are not seen. In ulcerated lesions, secondary inflammation may be present with admixed lymphocytes [189].
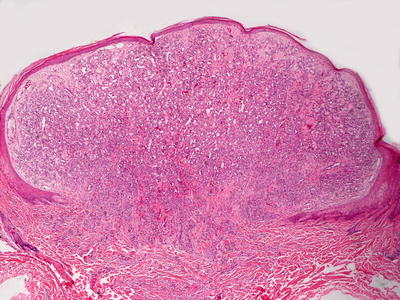
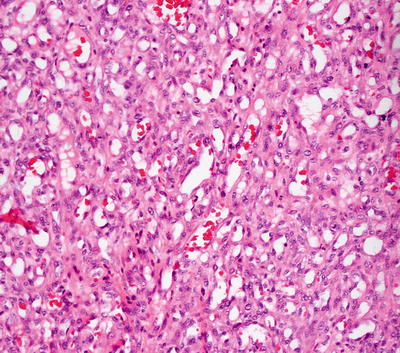
Fig. 21.24
Lobular capillary hemangioma (pyogenic granuloma ) demonstrates an exophytic proliferation of small, well-formed blood vessels surrounded by a proliferation of spindle-shaped cells
Fig. 21.25
Well-formed blood vessels interspersed with benign-appearing spindle-shaped cells and scattered mitoses are seen in lobular capillary hemangioma
The differential diagnosis includes an infantile hemangioma and bacillary angiomatosis. Infantile hemangiomas are differentiated based upon the clinical presentation , lobular configuration, and polypoid architecture. Bacillary angiomatosis can be differentiated based upon the clinical history of multiple lesions, and the presence of neutrophils and amorphous granular material (sheets of microorganisms) within the lesion. A congenital epithelioid angioma might enter into the differential diagnosis, but the presence of endothelial cell atypia is not expected in pyogenic granuloma [190]. Pyogenic granulomas can develop within underlying vascular lesions, such as port-wine stains, in which case histologic features of both processes can be seen [191, 192].
21.6.3.3 Pathogenesis
Pyogenic granuloma is thought to be related to prior trauma and drug reaction [193, 194]. Molecular analysis by genome-wide transcriptional profiling of blood vessels from pyogenic granuloma reveals a gene signature unique to this entity that includes genes in the nitric oxide pathway, hypoxia-induced angiogenesis, and vascular injury [195]. A gene found to be highly associated with pyogenic granuloma is FLT4 (vascular endothelial growth factor receptor-3 (VEGFR-3)), which is a receptor tyrosine kinase important in lymphangiogenesis. There is increased expression of vascular endothelial growth factor (VEGF) , transcription factors pATF2 and pSTAT3, as well as MAPK signal transduction in pyogenic granuloma that can potentially act as proangiogenic regulators to induce angiogenesis [196–198]. Somatic activating mutations in RAS and B-RAF have also been identified in pyogenic granuloma [199, 200].
21.6.4 Targetoid Hemosiderotic Hemangioma (Hobnail Hemangioma)
21.6.4.1 Clinical Features
It was proposed that this entity be named “superficial hemosiderotic lymphovascular malformation” [201]. This is an uncommon cutaneous vascular lesion thought to be primarily lymphatic in nature. It usually occurs in young to middle-aged adults (median age is 30 years old) on the trunk and extremities. There are rare reports in children [201, 202]. Clinically, the lesion presents as a solitary violaceous papulonodule surrounded by a pale halo and an ecchymotic rim (Fig. 21.26). Lesions are sometimes tender, especially with pressure. They are usually small and amenable to surgical excision if symptomatic.
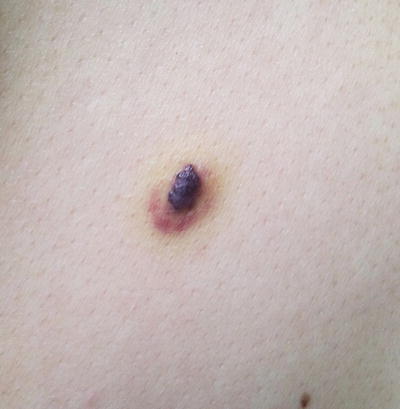
Fig. 21.26
Targetoid hemosiderotic hemangioma is a dark purple papule with surrounding ecchymotic patch (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
21.6.4.2 Histology
Histologic changes in targetoid hemosiderotic hemangioma include a poorly circumscribed vascular proliferation that is centered in the reticular dermis and extends into the subcutis. Dilated blood vessels demonstrate intraluminal papillary projections (Fig. 21.27). A single layer of endothelial cells protrudes into the vascular lumen in a pattern that resembles hobnails [203] (Fig. 21.28). Deeper angular and narrow vascular spaces are present around eccrine structures. The deep component dissects through collagen bundles in an invasive pattern, giving rise to an angiosarcoma-like growth pattern [204]. Mitotic activity is scant [204]. There are abundant extravasated erythrocytes with dermal hemosiderin deposition. Strong expression of the lymphatic marker podoplanin (D2-40) and lack of CD34 suggest that these lesions may be of lymphatic origin [205, 206].
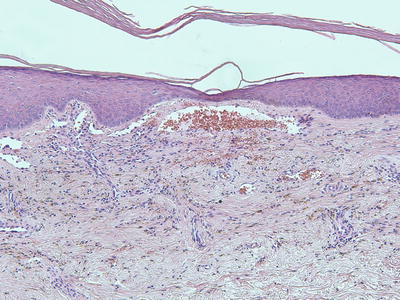
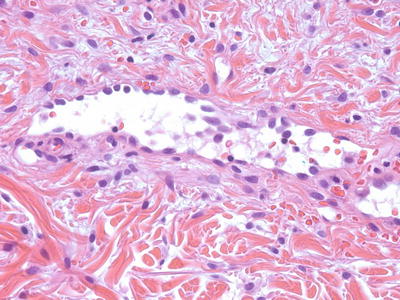
Fig. 21.27
Targetoid hemosiderotic hemangioma demonstrates diffusely arranged blood vessels that are occasionally slit-like and have surrounding hemosiderin
Fig. 21.28
Blood vessels in targetoid hemosiderotic hemangiomas are lined by endothelial cells that protrude into the lumen (hobnail endothelial cells)
The differential diagnosis includes angiosarcoma, Kaposi’s sarcoma, and retiform hemangioendothelioma [204]. Lack of cytologic atypia and mitotic activity helps to exclude angiosarcoma. Lack of a spindle cell proliferation, promontory sign and plasmacellular response argues against Kaposi’s sarcoma. Retiform hemangioendothelioma has a more retiform growth pattern that is not seen in targetoid hemosiderotic hemangioma. Epithelioid hemangioma and progressive lymphangioma may also enter the differential diagnosis. The invasive growth pattern would be unusual in an epithelioid hemangioma. The hobnail morphology characteristic of targetoid hemosiderotic hemangioma is not seen in progressive lymphangiomas .
21.6.4.3 Pathogenesis
It has been postulated that targetoid hemosiderotic hemangioma is a result of trauma [203, 207]. It has been proposed that obstruction of lymphatic vessels from trauma could result in inflammation, hemosiderin deposition, and fibrosis, which are histologic features seen in targetoid hemosiderotic hemangioma. Hormonal influence during the menstrual cycle can also potentiate the progression of this lesion [208].
Endothelial cells in targetoid hemosiderotic hemangioma express the lymphatic markers vascular endothelial growth factor receptor-3 (VEGFR-3) and podoplanin, indicating a lymphatic origin of this lesion [202, 203]. Moreover, this lesion has a low cell proliferative index by MIB-1 immunostain and no WT-1 expression, which is a molecular marker selectively expressed in vascular tumors but not in vascular malformations [102, 209]. These findings further indicate that this entity is a type of vascular malformation of lymphatic origin rather than a true vascular tumor [102, 202, 205].
21.6.5 Tufted Angioma
21.6.5.1 Clinical Features
Tufted angioma is a rare cutaneous vascular tumor. Approximately 25 % of lesions are present at birth, and 50 % become evident in the first year of life. Most lesions occur on the neck, shoulder, upper trunk and extremities [210]. Lesions often begin as erythematous macules or patches resembling a port-wine stain, and progress to larger purple/violaceous plaques or nodules over time [211] (Fig. 21.29). Some lesions are painful and tender. Acquired tufted angioma is also associated with a risk of coagulopathy or development of Kasabach–Merritt syndrome , which is a potentially fatal thrombocytopenic coagulopathy secondary to bleeding and platelet trapping within the lesion [212]. The mortality rate of Kasabach–Merritt syndrome has been estimated at 20 % [211, 213]. Some lesions improve or regress spontaneously over time. Smaller symptomatic lesions may be amenable to surgical excision .
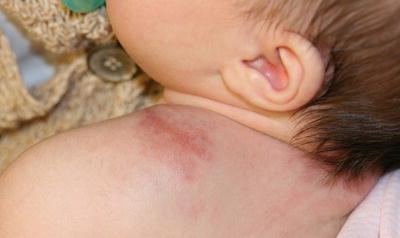
Fig. 21.29
Tufted angioma is a firm infiltrative mass with overlying telangiectasias on the shoulder of an infant
21.6.5.2 Histology
Tufted angioma, previously known as angioblastoma [214], demonstrates multiple lobules of blood vessels and surrounding cells within the reticular dermis and extending into the subcutis [215] (Figs. 21.30 and 21.31). The vessels are lined by endothelial cells and surrounded by pericytes. Mitotic activity is minimal, and there is no significant cytologic atypia in endothelial cells and surrounding pericytes. The tumor has a clear lobular growth pattern with foci of vascular proliferation, marked cellularity and interspersed zones of paucicellular dermis. The vascular spaces can be round or somewhat slit-like. As there is some tendency toward local recurrence and progressive growth, some authors consider these tumors to be of intermediate biologic behavior, although there are no known reports of distant metastasis [216]. A rare case of acquired tufted angioma occurring within an existing port-wine stain has been reported, and several of these lesions have arisen during pregnancy [191, 217].
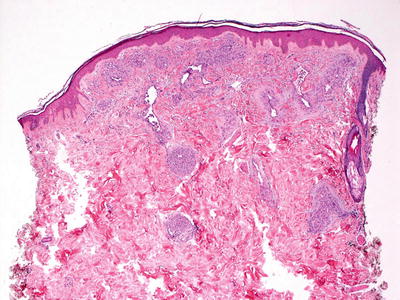
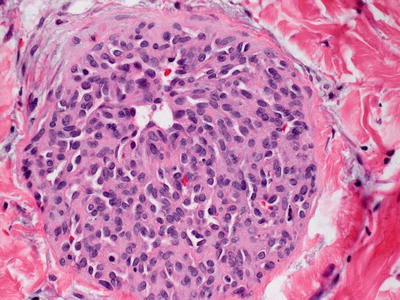
Fig. 21.30
Tufted angioma demonstrates a pattern of multiple lobules of vascular proliferation separated by normal collagen throughout the superficial and deep dermis
Fig. 21.31
Each of the cellular lobules in tufted angioma contains multiple small vessels surrounded by a proliferation of bland-appearing spindle-shaped cells
The differential diagnosis of tufted angioma includes Kaposi’s sarcoma due to the infiltrative growth pattern and the abundant cellularity beyond simple endothelial cells. Recognition of the central lobules of well-formed blood vessels as well as the clinical presentation should enable easy distinction in most cases. In addition, HHV-8 immunostain, which is positive in virtually all cases of Kaposi’s sarcoma , would be negative in acquired tufted angiomas [218]. Angiosarcomas also have an infiltrative growth pattern, but the proliferation is limited to endothelial cells that display marked cytologic atypia and mitosis [218].
21.6.5.3 Pathogenesis
Tufted angioma is a benign vascular tumor whose pathogenesis is unknown. Endothelial cells in tufted angioma express the lymphatic marker Prox-1, indicating their lymphatic origin or differentiation [219]. As has been noted previously, patients with tufted angioma may develop Kasabach–Merritt syndrome, which is a potentially life-threatening condition of thrombocytopenia, coagulopathy, and purpura. It has been postulated that the abnormal and convoluted vascular architecture in tufted angioma results in turbulent blood flow, thus promoting platelet trapping, platelet activation, and consumptive coagulopathy within the abnormal vasculature [212].
21.6.6 Kaposiform Hemangioendothelioma
21.6.6.1 Clinical Features
Kaposiform hemangioendothelioma (KHE) is a locally aggressive vascular neoplasm according to the current International Society for the Study of Vascular Anomalies (ISSVA) classification of vascular anomalies [56]. It mainly affects infants and children, although it may occasionally present in adulthood [220, 221]. KHE usually presents as firm violaceous nodules or plaques on the extremities or upper trunk (Fig. 21.32) [220, 222]. Rarely, they may involve visceral organs or bone
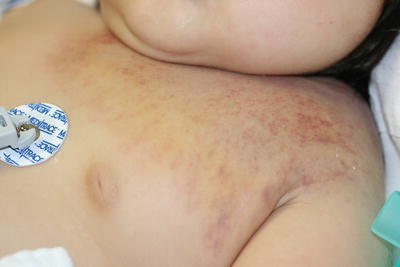
Fig. 21.32
A firm mass with overlying telangiectasias suddenly enlarged and became painful in a child with Kasabach–Merritt syndrome due to a kaposiform hemangioendothelioma of the chest
Similar to tufted angioma, KHE can be associated with Kasabach–Merritt syndrome [212]. Patients with Kasabach–Merritt syndrome typically present with a rapidly enlarging tumor mass in the setting of severe thrombocytopenia and coagulopathy. Disease management depends on the location and extent of the lesion. Although surgical excision is the definitive treatment, the location and large size of many lesions precludes complete resection. Medical treatment has historically included systemic steroids, interferon, and chemotherapeutic agents such as vincristine. Recent studies have provided evidence that rapamycin (sirolimus), an inhibitor of the mammalian target of rapamycin , is a new effective therapy for KHE [223, 224].
21.6.6.2 Histology
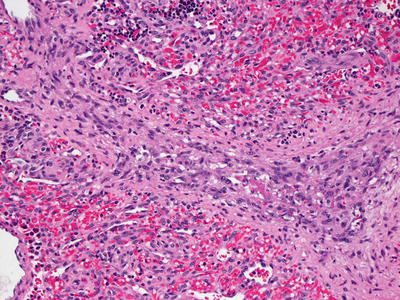
KHE is comprised of diffuse nodular proliferations of slit-like vessels surrounded by endothelial cells (Figs. 21.33 and 21.34). In some areas, the vessels are well formed, whereas in other areas, the vessels are poorly formed and have an infiltrative growth pattern, strongly mimicking Kaposi’s sarcoma [222]. Areas of dense cellularity comprised of endothelial cells and pericytes, and no apparent vascular differentiation may also be encountered. These areas may have a glomeruloid growth pattern [225]. Cytologic atypia is slight, and mitoses are infrequent and without atypical forms [226] (Fig. 21.35). Hemosiderin and hemorrhage are present in some cases [226]. These tumors occupy the entire dermis and can extend into the subcutaneous fat. Lateral and deep to the central tumor, large malformed lymphatic channels are present in more than half of the described cases [222, 225, 226]. Immunostaining shows that the neoplastic cell population consists of lymphatic endothelial cells as evidenced by focal staining with the lymphatic marker D2-40 [225](Figs. 21.36 and 21.37). KHE is often associated with Kasabach–Merritt syndrome, and extensive intravascular thrombus formation is observed in such cases [222]. The tumor has a propensity for local recurrence and persistence, although distant metastasis appears to be uncommon [216, 225, 227].
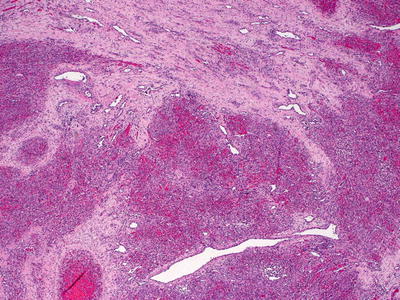
Fig. 21.33
A lobular configuration of dense cellularity and vascular differentiation characterizes KHE