, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
24.1 Pilomatricoma
24.1.1 Clinical Features
Pilomatricomas are common benign hair matrix tumors most frequently seen in individuals less than 20 years old [1]. Incidence of pilomatricomas is sporadic, although reports of heritable forms have been described with multiple lesions appearing in association with myotonic dystrophy, Gardner syndrome , Rubinstein–Taybi syndrome , and trisomy 9 [2].
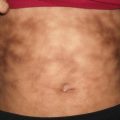
Pilomatricomas usually present as a solitary, mobile, firm, skin-colored to whitish subcutaneous nodules, occasionally with overlying bluish red discoloration (Fig. 24.1). In cystic variants, rapid enlargement with more pronounced erythema can occur secondary to hemorrhage. Overlying bulla can be seen in rare incidences. This bullous subtype is more common on the shoulders and upper extremities of females [3]. Surgical excision is curative, and it is generally recommended as lesions are at risk for inflammation secondary to trauma or infection. Potential for malignant transformation has been suggested, but this remains controversial with low reported incidence [4, 5].
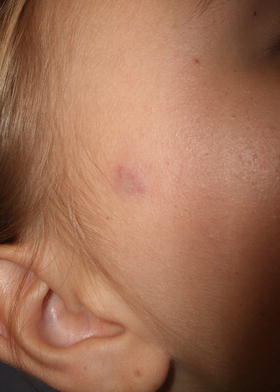
Fig. 24.1
Pilomatricoma appears as a firm nodule on the cheek of a child with overlying red and blue discoloration
24.1.2 Histology
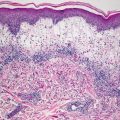
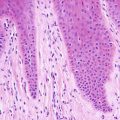
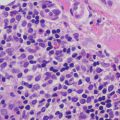
Biopsy of pilomatricoma demonstrates a well-circumscribed proliferation of basaloid cells giving rise to a central cavity comprised of “shadow” or “ghost cells” (Fig. 24.2). Germinative follicular basaloid cells rim the space and undergo abrupt transition to central squamous cells that lose their nuclei and are pink in color (Fig. 24.3). There is a tendency for the development of calcification in the central portion of the lesion (hence, the old name calcifying pilomatricoma of Malherbe). In cases where there is disruption of the intact cystic architecture, there is a marked granulomatous inflammatory infiltrate with multinucleated giant cells, keratin debris, and additional foci of calcification.
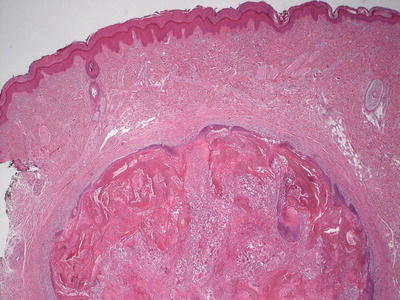
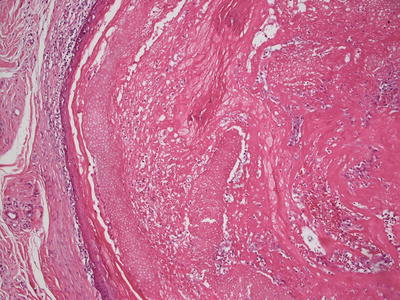
Fig. 24.2
Pilomatricoma demonstrates a sharply circumscribed dermal tumor consisting of basaloid cells at the periphery, ghost cells and a keratinaceous center
Fig. 24.3
Ghost cells and keratinocytes with no residual nuclei are the characteristic cells seen in pilomatricoma
The outermost portions of the cystic proliferation contain cells that are quite immature in appearance, and abundant mitotic figures are present. If only this portion of the lesion is available for examination, the tumor may resemble an undifferentiated malignant process. However, identification of the portion with “ghost” cells makes the diagnosis rather straightforward. The issue of tissue sampling is more a problem with fine needle aspiration specimens, as most punch or shave biopsies will contain both major elements of this neoplasm [6, 7]. Malignant transformation of these tumors can occur, but it is usually in long-standing, large lesions occurring in elderly patients, and these are not ordinarily encountered in children.
24.1.3 Pathogenesis
Pilomatricoma commonly carries somatic mutations in Bcl-2 (a gene that regulates cell apoptosis) and CTNNB1 (a gene that encodes β-catenin, which is important in hair follicle development) [8–10]. Pilomatricoma often occurs sporadically. However, there are reports of cases of multiple familial pilomatricomas as well as cases of pilomatricomas that occur in association with autosomal dominant conditions, such as myotonic dystrophy and familial adenomatous polyposis, and with syndromic conditions, such as Rubinstein–Taybi syndrome , Gardner syndrome , and Turner syndrome [11–13].
24.2 Trichoepithelioma
24.2.1 Clinical Features
Multiple trichoepitheliomas can be inherited in an autosomal dominant manner, appearing in early adulthood or less commonly in childhood [14]. Sporadic nonheritable occurrences of solitary lesions typically develop during the second and third decades of life [14]. Trichoepithelioma can present as solitary or multiple lesions, and it can be associated with multiple familial trichoepitheliomas, Brooke–Spiegler syndrome , Rasmussen syndrome , and Rombo syndrome [15–17].
Trichoepitheliomas present as small (2–5 mm), firm, waxy, skin-colored papules and papulonodules on the central face, specifically along the nose, forehead, upper lip, and eyelids (Fig. 24.4) [14]. Lesions can also be found at the scalp, neck, trunk, and perianal skin. Discrete papules may aggregate to form nodular plaques. Larger lesions may have telangiectasias along the surface. Trichoepitheliomas are benign but they may be removed for cosmetic or confirmatory purposes.
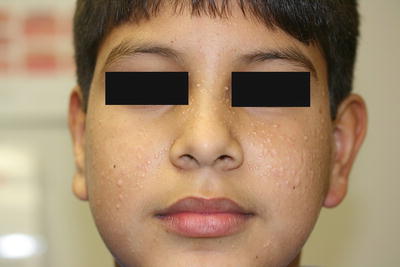
Fig. 24.4
Trichoepitheliomas present as multiple small, skin-colored papules over the face
24.2.2 Histology
Histologic findings of trichoepithelioma demonstrate a dermal tumor comprised of basaloid cells with follicular differentiation (Fig. 24.5). Connection to the overlying epidermis is unusual. Nests of basaloid cells are present surrounded by cellular eosinophilic stroma. The epithelial nests demonstrate varying degrees of follicular differentiation, with some foci being entirely basaloid cells, and others demonstrating full maturation with cystic cavities lined by granulated keratinocytes and containing hair follicles. In some nests, papillary mesenchymal bodies , which are in-pouchings of densely cellular mesenchymal cells resembling dermal papillae, are observed. It is common to see foci of granulomatous inflammation with abundant giant cells in areas where the keratin-filled cystic spaces have ruptured (Fig. 24.6). A variant of trichoepithelioma with sclerotic stroma and compressed thin strands of follicular basaloid cells is known as desmoplastic trichoepithelioma [18, 19]. Rarely, trichoepithelioma may have intradermal melanocytic nevus as part of the lesion. It remains unclear if this is a single neoplasm or coexistence of two common tumors [20].
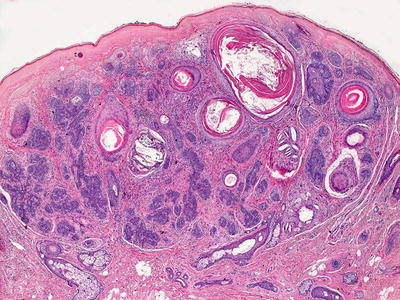
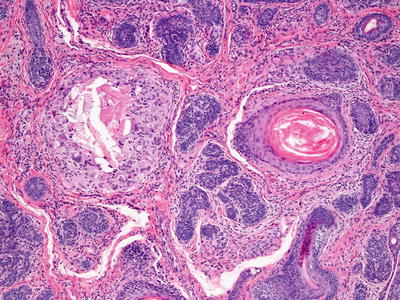
Fig. 24.5
Trichoepithelioma is characterized by nests of basaloid epithelium with follicular differentiation coursing through the dermis
Fig. 24.6
Trichoepithelioma demonstrates follicular structures, basaloid nests, and foci with granulomatous inflammation surrounding ruptured cysts
The main differential diagnosis is basal cell carcinoma, although these tumors are not common in children. Trichoepitheliomas lack the classic cleft formation between the epithelial and stromal components that are usually found in basal cell carcinoma. In addition, the stroma in basal cell carcinomas is more densely cellular and contains mucinous ground substance not seen in trichoepithelioma [18–20]. Papillary mesenchymal bodies are quite rare in basal cell carcinoma, which has more peripheral palisading of tumor cells at the edges of epithelial lobules [21]. Desmoplastic trichoepithelioma may also be confused with microcystic adnexal carcinoma, although this neoplasm is distinctly uncommon in children. In addition, no ductule differentiation is seen in desmoplastic trichoepitheliomas.
Trichoadenoma and trichoblastoma may share some histologic features with trichoepithelioma, but the distinction is of academic interest only. These are closely related benign follicular neoplasms. Trichoadenomas have less basaloid component and more complete follicular maturation than trichoepitheliomas. Trichoblastomas demonstrate more undifferentiated basaloid component and less follicular maturation than trichoepitheliomas .
24.2.3 Pathogenesis
Trichoepithelioma is a benign skin tumor of follicular germinative cells [22]. The patched gene (PTCH) appears to play an important role in the pathogenesis of trichoepithelioma. Frameshift and in-frame somatic deletions of PTCH as well as overexpression of PTCH mRNA have been identified in trichoepithelioma [23]. Brooke–Spiegler syndrome (BSS) is an autosomal dominant syndrome with adnexal tumors, including cylindromas, spiradenomas, and trichoepitheliomas. Germline mutations in the cylindromatosis (CYLD) gene have been identified in BSS [17, 24, 25]. CYLD is a deubiquitinating enzyme that negatively regulates the NF-κB and c-Jun N-terminal kinase pathways [26].
24.3 Trichofolliculoma
24.3.1 Clinical Features
Trichofolliculoma usually appears in adulthood [27]. It is a benign asymptomatic and solitary papule or nodule on the head or neck with greatest frequency on the face. Less common sites of involvement include the lip, nose, external auditory meatus, genitals, and extremities. There are very few cases of multiple lesions in the literature [27].
24.3.2 Histology
Trichofolliculoma appears as a single invagination at the point of a pilosebaceous unit. On rare occasions, more than one such invagination may occur. The central follicular cavity is slightly dilated. It is a fully mature hair follicle. Emanating from the sides of the dilated follicle are multiple follicular buds comprised of basaloid follicular epithelium with varying degrees of differentiation (Figs. 24.7 and 24.8). Vellus hairs may be present within these small basaloid buds. These may appear to attach to the central cavity or to be separate, although they do attach on other tissue planes of sectioning. These secondary follicular units occasionally give rise to tertiary follicular units. Surrounding the epithelial constellation is a cellular and eosinophilic stroma that separates the neoplasm from the surrounding dermis. Follicles may be in anagen, catagen, or telogen phase [28]. Mitotic activity is not abundant and atypical forms are not seen. The tumor is well circumscribed and has benign cytologic features. A rare variant occurs that contains sebaceous glands as part of the follicular growth. This tumor is known as a sebaceous trichofolliculoma [29, 30].
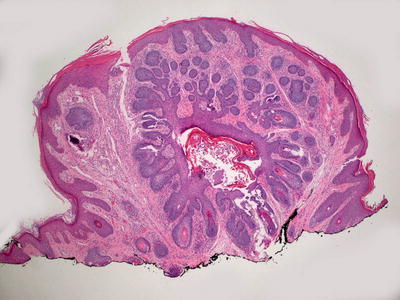
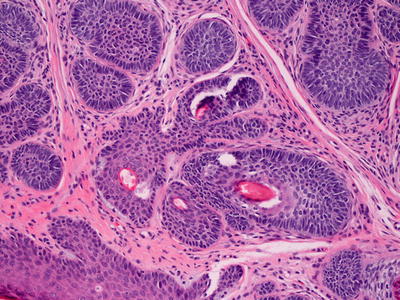
Fig. 24.7
A central hair follicle gives rise to abundant basaloid and primitive follicular structures in trichofolliculoma
Fig. 24.8
Basaloid buds and primitive follicular structures characterize trichofolliculoma
24.3.3 Pathogenesis
Trichofolliculoma is a benign hair follicle hamartoma. It is thought to form from an abortive differentiation of hair follicle stem cells towards a fully developed hair follicle during the hair cycle. Histological and immunohistochemical studies suggest that in trichofolliculoma, cytokeratin 15-positive hair follicle stem cells proliferate and form abnormal secondary hair follicles from a primary infundibulo-follicular primordium [31]. There are also abnormalities in the hair cycle and the regulation of hair follicle size in trichofolliculoma .
24.4 Chondroid Syringoma
24.4.1 Clinical Features
Chondroid syringoma presents as a nondescript slow-growing, asymptomatic, well-demarcated subcutaneous nodule, most frequently on the head or neck [32]. Occurrence of chondroid syringoma is rare with reported incidence of 0.01–0.02 % [32, 33]. Tumors are typically benign, but malignant transformation has been described.
24.4.2 Histology
Histologic features of chondroid syringoma include a dermal-based neoplasm comprised of tubular and ductular epithelium coursing in a myxoid stroma (Fig. 24.9). There is great variation in the proportions of stroma and epithelial components. Some tumors have abundant myxoid or mucinous stroma that bears histologic resemblance to cartilage. Others have more fibrous stroma with only scant foci of myxoid change. The epithelial component varies from cases with small glandular structures lined by cuboidal epithelium and underlying myoepithelium to cases with interanastamosing tubular structures (Fig. 24.10). The tubular structures are similarly lined by cuboidal epithelium, and in most cases, myoepithelium is present as well. These have been designated as eccrine and apocrine types of chondroid syringomas, respectively [34]. While this distinction helps to describe the varying histologic patterns and perhaps pathogenesis, there is no difference in biologic behavior, and the separation is of academic interest only. In some cases, the tumors consist solely of myoepithelial cells and are called myoepitheliomas, bearing some resemblance to similarly named salivary gland neoplasms [35]. While a malignant counterpart of chondroid syringoma sometimes occurs, this benign tumor is exceedingly uncommon in children, and malignant lesions have not been described in children [36].
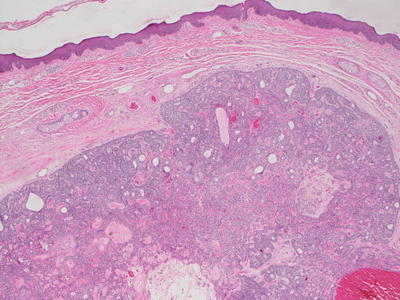
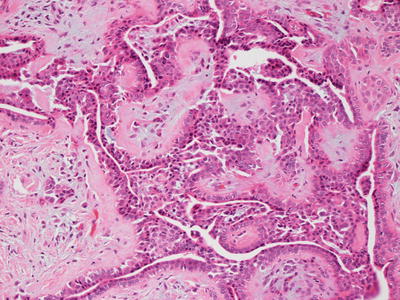
Fig. 24.9
Chondroid syringoma is a well-circumscribed dermal tumor with glandular structures surrounded by small blue cells
Fig. 24.10
Glandular structures and tubules interanastamose in a mucinous stroma in chondroid syringoma
24.4.3 Pathogenesis
Chondroid syringoma is a tumor of epithelial origin with the capacity to form adnexal structures and produce chondroid matrix [37]. It can have apocrine or eccrine differentiation with frequent occurrence of folliculo-sebaceous differentiation in the apocrine type of chondroid syringoma [34]. Merkel cells have been identified in this tumor. However, the clinical significance of the presence of Merkel cells in this lesion is unknown [38].
24.5 Myoepithelioma
24.5.1 Clinical Features
Cutaneous myoepithelioma is an uncommon and relatively recently identified tumor. In one cohort of patients, the median age of onset was 39 years old with a range of age presentation between 2 months and 74 years old. Another retrospective review suggested peak incidence in childhood with a younger median age of 22 years old [39, 40]. Slight male predominance was noted in both studies. Lesions are commonly described as slow-growing painless nodules on some aspect of an extremity, although tenderness, swelling, and erythema have been noted in at least one case [39]. Myoepitheliomas appear to follow a benign course, but local recurrence may occur following incomplete excision [40].
24.5.2 Histology
Closely related to mixed tumors (chondroid tumors), myoepitheliomas are composed entirely of myoepithelial cells [35]. The tumors are sharply circumscribed and the stroma can be densely fibrotic or myxoid (Fig. 24.11) [41]. The cells grow in fascicles, and can be ovoid, spindly, or even histiocytoid (Fig. 24.12). A solid or cystic growth pattern is seen in most cases, but syncytial pattern has also been described [40]. The syncytial pattern shows cells with abundant eosinophilic cytoplasm and indistinct cellular borders. Cytologic atypia is minimal, and necrosis and vascular invasion are not found. In most cases, there is only slight pleomorphism [42]. The proliferation of myoepithelial cells can display a plexiform growth pattern [43]. Rare cases exist with dendritic or spindle-shaped melanocytes present within the lesions [44].
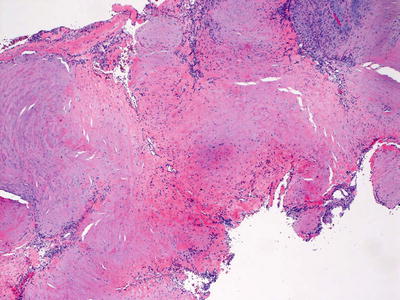
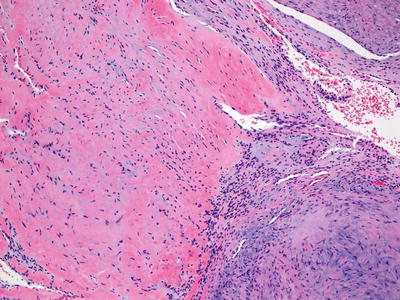
Fig. 24.11
A spindle cell proliferation is present within a background that is focally myxoid and fibrotic in myoepithelioma
Fig. 24.12
Bland spindle cells course within focally myxoid stroma in myoepithelioma
24.5.3 Pathogenesis
Cutaneous myoepitheliomas harbor EWSR1 gene rearrangement [45]. The frequency of EWSR1 rearrangement varies in these tumors, ranging from 29 to 44 % of the cases [45, 46]. Myoepithelial tumors with EWSR1 fusion affect children and young adults [40]. Documented fusion partners of EWSR1 include POU5F1, PBX1, and ZNF444 [46–48].
24.6 Eccrine Poroma
24.6.1 Clinical Features
It is estimated that sweat gland tumors represent approximately 1 % of cutaneous growths, and eccrine poroma accounts for 10 % of these cases with no reported gender or racial predilection [49]. Eccrine poromas typically occur in adults over 40 years old, although cases in children exist.
Eccrine poromas most commonly present as an asymptomatic, solitary nodule or papulonodule on the palm or sole, but they may occur on any area of the body that has a large number of eccrine sweat glands, including the fingers, trunk, head, and neck (Fig. 24.13) [49]. There are reports of porocarcinoma in long-standing lesions as well as recurrence of lesion following incomplete excision, thus making complete surgical excision the treatment of choice [49].
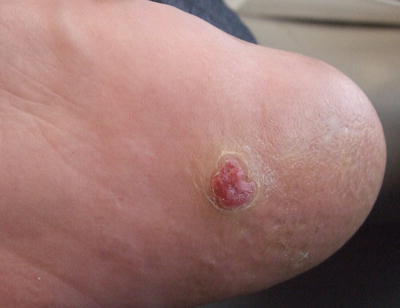
Fig. 24.13
An eccrine poroma appears as an exophytic, erythematous lobulated plaque with collarette of hyperkeratotic scale on the plantar foot
24.6.2 Histology
Eccrine poroma has sheets and cords of interanastamosing, bland cuboidal epithelium that grow down from the overlying epidermis (Fig. 24.14). The base of the lesion is flat, and it does not have an infiltrative deep border. Scattered ducts are present within the epidermis (Fig. 24.15). Cystic dilatations may be seen, and eosinophilic secretion is often present within them. Eosinophilic cuticles are apparent surrounding the ducts and cystic spaces. Rare mitoses are seen within the proliferations, but there is no tissue necrosis. Cytologic atypia is minimal. The underlying dermis contains abundant ectatic vessels, often located high within the papillary dermal tips.
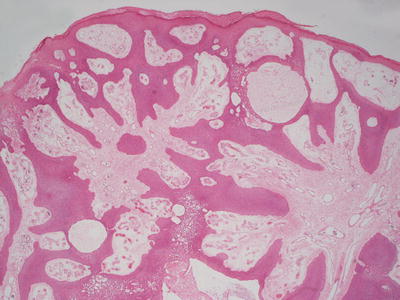
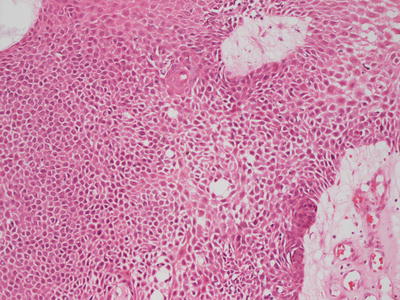
Fig. 24.14
Eccrine poroma is characterized by sheets of monomorphous cuboidal epithelium growing down from the epidermis in interanastamosing sheets
Fig. 24.15
Eccrine poroma demonstrates small glandular ducts within the sheets of monomorphous and benign-appearing epithelium
24.6.3 Pathogenesis
Eccrine poroma arises from the proliferation and expansion of basal cells of eccrine ducts. Comparative immunohistochemical studies of eccrine poroma and normal eccrine glands of the skin have shown that poroma cells have a similar immunophenotype to basal cells of eccrine ducts [52].
24.7 Papillary Eccrine Adenoma
24.7.1 Clinical Features
Papillary eccrine adenoma is an uncommon benign sweat gland tumor, most often occurring in young black women with an age range between 9 and 69 years old [53]. Papillary eccrine adenoma is described as 0.5–2 cm reddish brown or gray nodules or papules, most often presenting on the wrist or foot. There are no reports of metastasis, and there is low likelihood of recurrence with complete excision of the lesion [53].
24.7.2 Histology
Histologic changes of papillary eccrine adenoma are those of a well-circumscribed proliferation of dilated eccrine structures present in the dermis. The glandular structures are lined by one or several layers of cuboidal epithelium resembling eccrine structures [54]. There are abundant infoldings and papillomatous projections into the glandular lumen (Figs. 24.16 and 24.17). Each of the glandular units remains intact, and there is no invasion into the surrounding dermis. Cytologic atypia and mitotic activity are minimal. The surrounding stroma can be somewhat fibrotic [55]. There is great overlap between papillary eccrine adenoma and what has been called tubular apocrine adenoma. Some authors consider them to be synonymous. They may represent a spectrum of benign glandular neoplasms of cutaneous appendages that demonstrate varying degrees of apocrine differentiation [56]. The distinction is of academic interest only as these tumors have the same propensity to recur, but they do not metastasize [57].
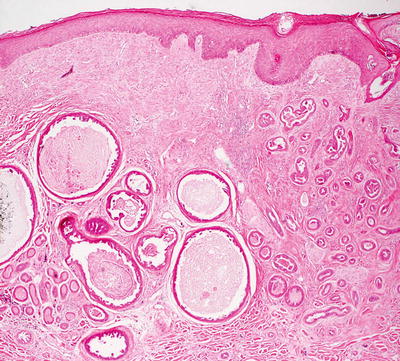
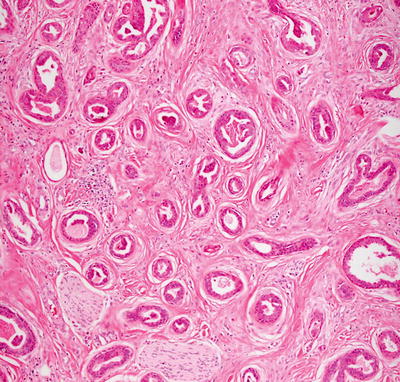
Fig. 24.16
Papillary eccrine adenoma demonstrates well-formed glands within the dermis, some of which contain papillary infoldings
Fig. 24.17
Dermal glandular structures are lined by cells with either eccrine or apocrine appearance in papillary eccrine adenoma
The differential diagnosis includes eccrine carcinoma. These tumors are quite rare in children and are differentiated from papillary eccrine adenoma based upon cytologic atypia, mitotic activity, foci of necrosis, and an invasive growth pattern .
24.8 Syringoma
24.8.1 Clinical Features
Syringomas occur most frequently in adolescence and early adulthood with greater prevalence among females [58]. Familial cases have been reported as well as increased incidence in certain patient populations, such as those with Down syndrome [58].
24.8.2 Histology
Histologic features of syringomas include a proliferation of well-formed tubular and ductular structures coursing in a dense fibrous stroma in the superficial portion of the dermis (Fig. 24.18). The dense stroma clearly delineates this benign proliferation from the surrounding uninvolved dermis, and it is a useful feature for establishing the diagnosis. Small tadpole-like or comma-like structures and other fully mature eccrine structures are present (Fig. 24.19). Two layers of cuboidal epithelium that resemble eccrine ductular cells line the structures. Cytologic atypia is not present, and mitotic activity is quite rare. A variant that is comprised of ductular lining cells with abundant clear cytoplasm, known as a clear cell syringoma, is found in some patients with diabetes [59, 60].
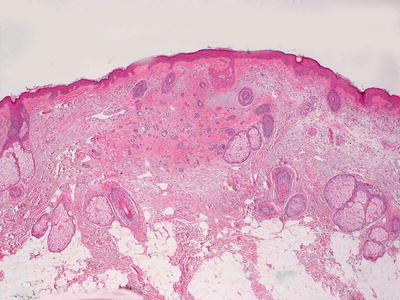
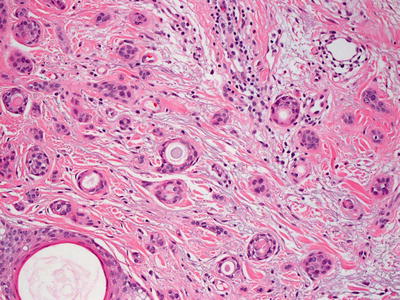
Fig. 24.18
Small glandular structures coursing in fibrous stroma are present in the superficial dermis in syringoma
Fig. 24.19
Tadpole-like configurations and well-formed ductules are seen in syringoma
The major differential diagnosis of syringoma is microcystic adnexal carcinoma (MAC), which is a tumor that essentially does not occur in children. MAC differs from syringoma in that it is not sharply circumscribed but has a deeply invasive growth pattern with perineural invasion. There is also biphasic differentiation with a follicular component in MAC. While cytologic atypia and mitotic activity are not seen in MAC, the deep invasion and lack of circumscription make histologic distinction possible. Desmoplastic trichoepithelioma may also enter the differential diagnosis, but it is distinguished based upon cystic structures with granular layers and keratin-filled centers as opposed to the ductular structures seen in syringoma .
24.9 Hidradenoma Papilliferum
24.9.1 Clinical Features
Hidradenoma papilliferum often occurs in adult females [61]. This lesion has nondescript clinical features and is characterized as a solitary subcutaneous nodule, occasionally with a cystic quality that may contain serous material. Hidradenoma papilliferum specifically affects anogenital mammary-like glands in the vulva and perianal skin [61]. The lesion is benign but may be surgically excised if desired.
24.9.2 Histology
Hidradenoma papilliferum occurs most frequently on the vulva, but it has been described in many other sites [62]. The usual histologic presentation is that of a sharply circumscribed dermal-based tumor with no connection to the overlying epidermis. However, invagination down from the epidermis has been observed in some cases [63, 64]. The tumor consists of epithelial-lined papillomatous growths with focal cystic spaces (Figs. 24.20 and 24.21). The lining is composed of cuboidal or columnar-type epithelium with focal subjacent myoepithelial cells. Mitoses can be found within the epithelial lining, but are not predictive of biologic behavior [65]. Oxyphilic or apocrine metaplasia, characterized by marked eosinophilia within the lining epithelial cells, occurs in some cases [66]. Clear cell metaplasia of the myoepithelial layer has also been described [66]. The intervening stroma is fibrous, but unlike syringocystadenoma papilliferum, which shares some histologic characteristics, plasma cells do not fill the surrounding dermis. Recent literature suggests that these tumors arise from mammary-like anogenital glands [67]. Fine needle aspiration biopsy is not the standard procedure for establishing the diagnosis, but it has been described as effective [68].
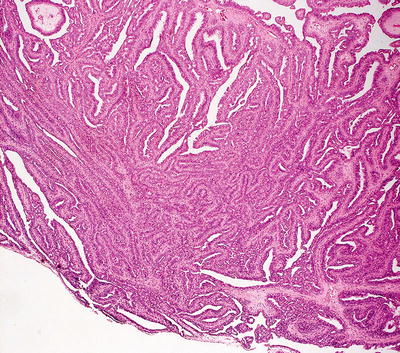
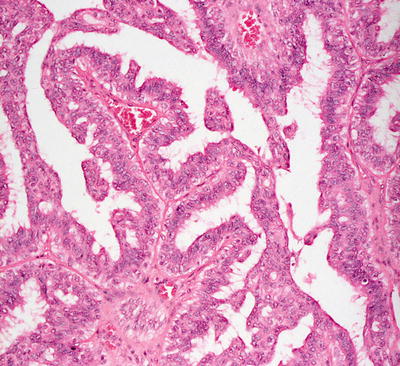
Fig. 24.20
Glandular epithelium with a papillomatous architecture is seen in hidradenoma papilliferum
Fig. 24.21
Apocrine-type epithelium with hobnailing into the lumen is seen lining the papillomatous spaces in hidradenoma papilliferum
Malignant transformation of hidradenoma papilliferum is exceedingly uncommon [61]. No cases have been described in children. Several cases, including one that underwent malignant transformation, have been associated with human papillomavirus 16, but this association remains unclear [61, 66].
The histologic differential diagnosis includes syringocystadenoma papilliferum which is distinguished primarily based upon its anatomic site with a predilection for the scalp and forehead. Other distinguishing features include the connection to the epidermis and plasma cell infiltrate. These features are present in syringocystadenoma papilliferum but are not ordinarily seen in hidradenoma papilliferum. Metastatic carcinoma might also enter the differential diagnosis, but the sharp circumscription, lack of significant cytologic atypia, relative paucity of mitoses in most cases, and lack of necrosis all favor a diagnosis of hidradenoma papilliferum .
24.10 Syringocystadenoma Papilliferum
24.10.1 Clinical Features
More than half of syringocystadenoma papilliferum are present at birth with an additional 13–15 % of cases presenting during childhood [69]. Up to 40 % of cases occur within a nevus sebaceous, while the remainder present de novo.
Syringocystadenoma papilliferum commonly occurs on the scalp and forehead. The clinical appearance of this tumor is variable. The surface may be smooth or verrucous [69]. Syringocystadenoma papilliferum is most commonly described as a 1–4 cm solitary plaque with overlying alopecia. Occasionally the plaque may be comprised of discrete small papules in a linear configuration with central invagination, reminiscent of molluscum contagiosum. The surface may become more mammillated and exophytic during puberty or pregnancy. Lesions may ulcerate with subsequent crusting. Very rarely, malignant transformation may occur in later life with a few reports of syringocystadenocarcinoma occurring in adults [69].
24.10.2 Histology
Syringocystadenoma papilliferum invaginates down from an otherwise unremarkable epidermis (Fig. 24.22). Two layers of cells line the cystic invagination. The layer closest to the cystic cavity is a layer of columnar cells that are eosinophilic and occasionally demonstrate apocrine-type decapitation secretion. Deep to this layer of cells is a layer of flattened myoepithelial cells. The superficial portion of the invagination may be lined with epithelial cells that have undergone squamous metaplasia [70]. Emanating from the cystic space are papillary infoldings of the epithelial lining (Fig. 24.23). As they extend deep to the invagination, these papillomatous infoldings may give rise to apocrine cystadenomas [71]. Deep to the lining is a brisk inflammatory infiltrate that is composed primarily of plasma cells.
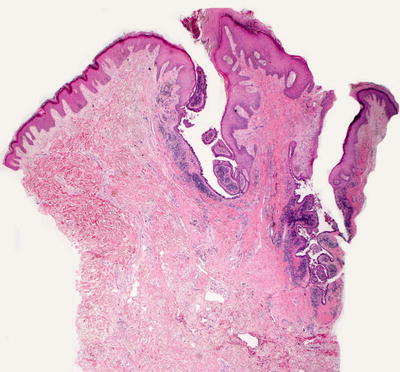
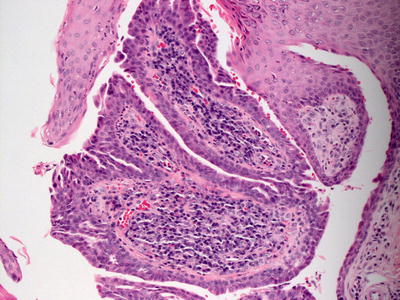
Fig. 24.22
Syringocystadenoma papilliferum demonstrates an invagination down from the surface epidermis and is lined by glandular epithelium
Fig. 24.23
A papillomatous growth pattern lined by glandular epithelium and a dense underlying plasmacellular infiltrate are seen in syringocystadenoma papilliferum
24.10.3 Pathogenesis
Syringocystadenoma papilliferum harbors HRAS mutations, which are present in 26 % of cases of sporadic tumors [74]. The HRAS hotspot mutation (c.37GNC) is the predominant mutation in this lesion. In addition to HRAS mutations, BRAF V600E mutation has been found in 50 % of sporadic cases [74, 75]. These findings indicate that activating mutations in HRAS and BRAF contribute to the tumorigenesis of syringocystadenoma papilliferum. Other studies have suggested that p16 may also be a relevant tumor suppressor gene in the development of this tumor [76].
24.11 Tubular Apocrine Adenoma
24.11.1 Clinical Features
Tubular apocrine adenoma is a rare sweat gland neoplasm most often occurring on the scalp, although reports exist of tumors at the lower extremities, back, and genitals, including a case presenting on the vaginal introitus of an 8-year-old girl [77]. The tumors are described as a slow-growing nodule or papule that may be darkly pigmented or erythematous [77, 78]. The lesions are benign with no demonstrated potential for metastasis, but they can be locally invasive.
24.11.2 Histology
Histologic findings of tubular apocrine adenoma are identical to those described in papillary eccrine adenoma (see Sect. 24.7), and most authors believe these entities to be identical (Figs. 24.24 and 24.25) [54, 56]. Others find slight histologic distinction of no biologic significance [55].
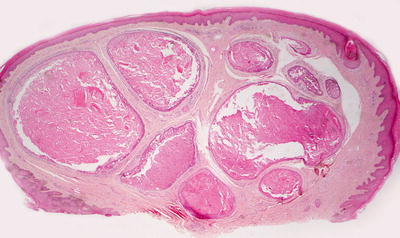
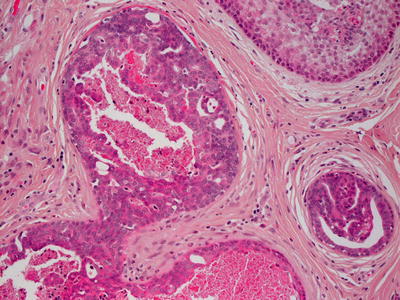
Fig. 24.24
Tubular apocrine adenoma demonstrates well-formed glands with papillary infoldings within the dermis
Fig. 24.25
Apocrine-type epithelium lines the papillary infoldings in tubular apocrine adenoma
24.12 Sebaceous Hyperplasia
24.12.1 Clinical Features
Sebaceous hyperplasia typically occurs in individuals over 40 years old [79]. Premature sebaceous hyperplasia is defined as non-pathologic proliferation of sebaceous glands occurring during or immediately following puberty, although reports in infants also exist [80]. Functional familial sebaceous hyperplasia, which is considered by some authors to be a variant of nevus sebaceous of Jadassohn, often presents in pubertal children. No gender predilection has been identified.
Sebaceous hyperplasia is characterized by yellowish small papules, typically located on the forehead, cheeks, neck, and lips. Lesions may coalesce linearly or form Blaschkoid plaques [81]. Lesions also occur less frequently on the oral mucous membranes, vulva, chest, areola, and penile shaft. Sebaceous hyperplasia and visible sebaceous glands on the oral and genital mucosa are termed Fordyce spots. Functional familial sebaceous hyperplasia is characterized by a yellowish plaque with peau d’orange surface on the face, chest, and upper back, with sparing around the orbits, nose, and ears [79]. Treatment may be pursued for cosmetic purposes with lesions amenable to retinoids or various surgical therapies.
24.12.2 Histology
Histologic findings include enlarged clusters of mature sebocytes that have a single layer of basaloid cells with full maturation with progression into the center of each cluster of cells (Fig. 24.26). Mitotic activity is rare, and cytologic atypia is not present [82]. The frequency of sebaceous hyperplasia may be increased in individuals who have undergone organ transplantation [83]. Similar histologic changes, although with secondary inflammation and glandular rupture and distortion, are seen in neonates with acne neonatorum [84].
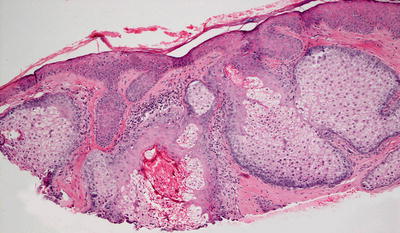
Fig. 24.26
Large and fully mature lobules of sebocytes are present in sebaceous hyperplasia
A Fordyce spot demonstrates the presence of ectopic sebaceous glands on either genital or oral mucosa without concomitant follicular units (Fig. 24.27) [85]. The normal appearing sebaceous glands are connected to the overlying epidermis by a duct. There is neither host immune response nor cytologic atypia in these lesions. In some cases, the glandular units may appear slightly hyperplastic. Recently, increased incidence of Fordyce spots has been recognized in patients with Lynch syndrome [86, 87].
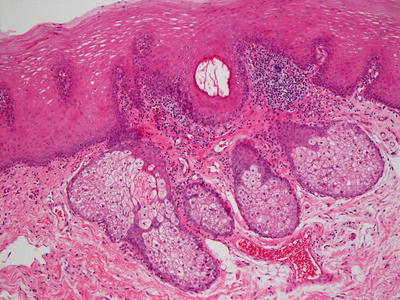
Fig. 24.27
Mature and fully developed sebaceous glands are seen beneath normal mucosa in Fordyce spot
24.12.3 Pathogenesis
Sebaceous gland hyperplasia is a benign hypertrophy of normal sebaceous glands. Sebaceous hyperplasia commonly occurs in adults with aging, but some cases in children have been reported. Sebaceous hyperplasia has been reported to occur in association with cyclosporine treatment, which is an immunosuppressive agent to prevent tissue rejection after solid organ transplantation [88]. It is present in 6–30 % of immunosuppressed organ transplant recipients as compared with 1 % of matched controls [83, 89, 90]. In these cases, the patients were receiving cyclosporine, suggesting that cyclosporine may be a causative factor in the development of sebaceous hyperplasia. Of note, this phenomenon has not been reported in children who received organ transplants. This may be due to the immaturity of the pilosebaceous unit in children [90].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


