, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
1.1 Atopic Dermatitis
1.1.1 Clinical Features
Atopic dermatitis is a multifactorial, primarily type 2 T-helper cell immune-mediated, inflammatory disorder characterized by chronicity, xerosis, and pruritus. Sixty percent of cases present within the first year of life, with 90 % of patients demonstrating clinical disease by 5 years old [1]. The estimated prevalence in industrialized nations, such as Europe and the USA, is 15–30 %, affecting about 25 % of children and 3 % of adults [1]. There is a strong genetic predisposition with 70 % of patients having a positive family history of atopy.
Clinical examination shows erythematous, xerotic, scaled patches and plaques with or without weeping crust and overlying excoriations. Lichenification and exaggerated skin markings highlight disease chronicity. In infants, eczematous lesions are found on the face and extensor surfaces with sparing of the diaper area, owing to pronounced humidity at that site. In contrast, older children and adults usually have skin findings localized to the flexural skin of the neck, antecubital fossae, wrists, popliteal fossae, dorsum of the feet and ankles (Fig. 1.1).
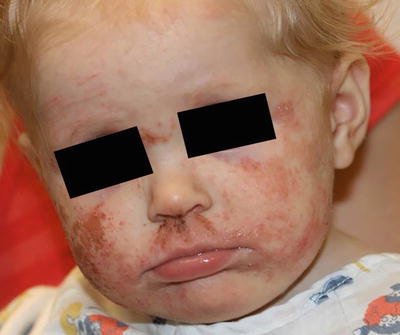
Fig. 1.1
Atopic dermatitis presents as erythematous, scaly plaques over the face of a toddler
Atopic dermatitis is described as the first “step” of the atopic march with 40–60 % of patients later developing allergic rhinoconjunctivitis and/or asthma [2]. Although atopic dermatitis has a relapsing course, most patients have resolution of disease by puberty with only 30 % having persistence into adulthood [1].
1.1.2 Histology
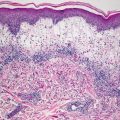
Histologic changes in acute atopic dermatitis are quite subtle. Scant wispy parakeratosis overlies an epidermis that demonstrates slight spongiosis and small collections of Langerhans cells [3] (Fig. 1.2). Scattered eosinophils may be present in the superficial dermis, admixed with increased numbers of lymphocytes [4] (Fig. 1.3). Degranulated mast cells may be present but are rarely observed on routine histologic sections. A folliculocentric pattern with spongiosis concentrated within follicular infundibula has been described, and is more common in darker skinned individuals [5].
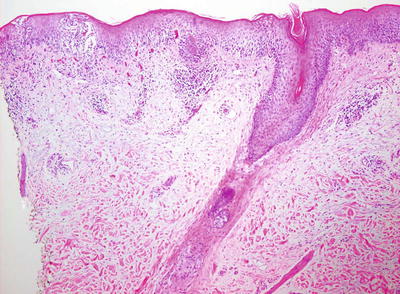
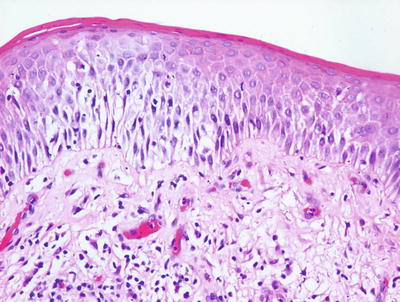
Fig. 1.2
Atopic dermatitis presents with a mild spongiotic dermatitis and a mild lymphocytic infiltrate in the superficial dermis
Fig. 1.3
A spongiotic dermatitis with scant exocytosis of lymphocytes is seen in atopic dermatitis
In the chronic phase, changes are nonspecific and are those of a chronic spongiotic dermatitis, often with superimposed lichen simplex chronicus-like changes. Hyperkeratosis is present diffusely overlying an acanthotic epidermis with elongated rete ridges and hypergranulosis. Spongiosis is only minimally present and there is a sparse perivascular lymphocytic infiltrate. Eosinophils are minimally increased in number. A specific diagnosis of chronic atopic dermatitis is not possible based solely upon histologic changes .
1.1.3 Pathogenesis
Genetics studies have revealed a strong association between atopic dermatitis and the epidermal barrier protein filaggrin , particularly in Northern Europeans [6–8]. Depending on the population, between 10 and 50 % of patients with atopic dermatitis carry filaggrin gene mutations [7, 9, 10]. In particular, two loss-of-function genetic variants of filaggrin (R510X and 2282del4) are strong predisposing factors for atopic dermatitis [7, 8, 11]. These variants also show significant association with asthma in the setting of atopic dermatitis.
The filaggrin gene is located within the epidermal differentiation complex, which is a large cluster of genes (including filaggrin, loricrin, and involucrin) that are important in keratinocyte terminal differentiation [8, 11]. The gene product is profilaggrin, which is a major component of the keratohyaline granules in keratinocytes. Profilaggrin –>is proteolyzed to produce functional filaggrin monomers. These monomers coalesce to form a dense protein–lipid matrix of the cornified cell envelope. This structure serves an epithelial barrier function that prevents epidermal water loss and impedes the entry of allergens, chemicals, and microorganisms across the skin barrier [12, 13]. Although filaggrin mutation is a major predisposing factor for classic atopic dermatitis, many patients with atopic dermatitis do not have defects in filaggrin. Thus, other genetic factors that influence skin barrier function may also be important. Potential candidate molecules are proteins that are involved in the processing of profilaggrin, such as retroviral-like aspartic protease (SASPase), caspase-14 and bleomycin hydrolase [14].
The antimicrobial skin barrier is compromised in atopic dermatitis, leading to secondary bacterial infection, such as Staphylococcus aureus colonization that can exacerbate inflammation in the disease process [15]. This is consistent with the clinical findings of impetiginization, folliculitis, and even skin abscesses as complications of infection in atopic dermatitis. The microbial exacerbation may be due to the augmentation of IgE production that is directed towards staphylococcal exotoxins [6]. Toll-like receptors (TLRs) are important in the detection of invading pathogens. There is evidence of impairment of toll-like receptor-2 (TLR2) expression and function in monocytes and macrophages in atopic dermatitis [16, 17]. TLR2 polymorphism (R753Q) has been reported in patients with severe form of atopic dermatitis, but the role of this polymorphism remains controversial [18, 19].
T lymphocytes play important roles in the pathogenesis of atopic dermatitis. The important T lymphocyte subsets in this disorder are type 2 T-helper cells (Th2) , type 17 T-helper cells (Th17) , and type 22 T-helper cells (Th22) [20–22]. The immune infiltrates in chronic atopic dermatitis skin are mainly composed of Th2 and Th22 cells, and some Th17 cells [22]. Cytokines produced by Th2 cells, particularly IL-4, IL-13, and IL-31, regulate important functions in the epidermal barrier, including epidermal cornification and the production of antimicrobial peptides [9]. Th2 cells also produce cytokines and interferon-gamma (IFN-gamma) that can affect filaggrin expression and epidermal differentiation [23]. Th17 cells produce IL-17 and some IL-22, and they regulate the production of antimicrobial peptides in keratinocytes as well as chemotaxis of neutrophils. IL-17 levels are decreased in atopic dermatitis skin, and that may contribute to the decreased production of antimicrobial peptides. Regulatory T cells (Tregs) are important in suppressing T-cell mediated immune response [24]. Loss of Treg function has been reported in atopic dermatitis [25].
Vascular changes and angiogenesis are other key features in atopic dermatitis. High levels of vascular endothelial growth factor-A (VEGF-A) are present in the skin of atopic dermatitis patients and appear to correlate with disease activity [26–28]. Activated macrophages can induce VEGF expression and angiogenesis during inflammation and contribute to the disease process.
Atopic dermatitis can occur as part of a syndrome, such as Netherton syndrome which is characterized by ichthyosis, chronic dermatitis, asthma, and allergic rhinitis [29]. Netherton syndrome is caused by mutations in the SPINK5 (serine protease inhibitor Kazal-type 5 ) gene, which encodes the protease inhibitor lymphoepithelial Kazal-type-related inhibitor (LEKTI) [30, 31]. Genetic studies have demonstrated linkage between polymorphisms in SPINK5 gene in Netherton syndrome and atopic dermatitis, asthma, and high levels of serum IgE [31, 32].
1.2 Dyshidrotic Eczema
1.2.1 Clinical Features
Dyshidrotic eczema, also known as pompholyx, is a chronic relapsing, vesiculobullous disorder of the palms and soles (Fig. 1.4). It is seen worldwide, though perhaps less frequently in Asian populations [33]. Associated conditions include both atopic dermatitis and contact dermatitis, with contact sensitivity noted in approximately one-third of affected individuals [33].
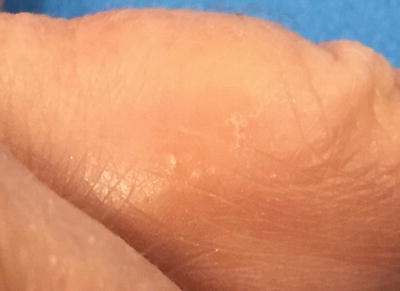
Fig. 1.4
Dyshidrotic eczema presents as scaly papules and deep-seated vesicles on the lateral finger
On physical exam, there are tense, deep-seated, “tapioca-like” vesicles as well as larger bullae on the palms, soles, and lateral fingers. Erythema and scaling may or may not be present. Involvement is usually bilateral. Patients may complain of pruritus or a burning sensation, and hyperhidrosis is a frequent feature, typically with increased occurrence during warmer seasons and climates [33]. Management hinges on avoidance of identifiable triggers and treatment of relapses of eczematous flares.
1.2.2 Histology
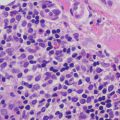
Histologic features of dyshidrotic eczema are nonspecific. The only distinguishing feature from other spongiotic processes is the acral location for this eruption (Fig. 1.5). Parakeratosis is present (except in very acute lesions) overlying an irregularly acanthotic epidermis with focal spongiosis that can range from slight to marked changes [34]. Collections of Langerhans cell microabscesses are seen in most cases (Fig. 1.6). Within the dermis, there is an infiltrate of lymphocytes and eosinophils may be present. Late lesions may demonstrate only minimal spongiosis and are indistinguishable from lichen simplex chronicus. In most cases, a PAS stain is recommended to exclude acral fungal infection, as the histologic changes may be identical .
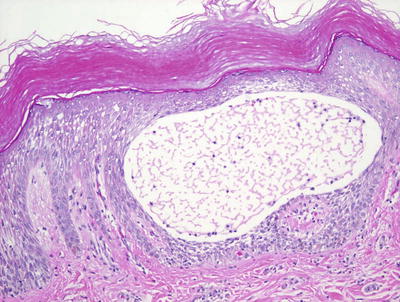
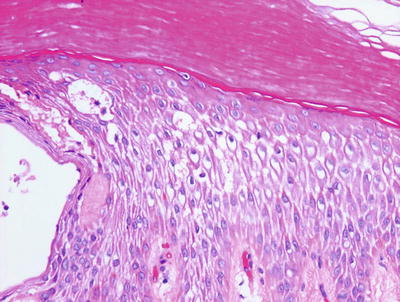
Fig. 1.5
Acral skin demonstrating spongiotic dermatitis with microvesiculation is present in dyshidrotic eczema
Fig. 1.6
Dyshidrotic eczema reveals a spongiotic dermatitis on acral skin
1.2.3 Pathogenesis
The pathogenesis of dyshidrotic eczema is not known. A number of factors, such as atopy, contact allergy, dermatophyte infection and drug eruption, can influence the development of dyshidrotic eczema in predisposed individuals. Studies of a large Chinese family with a rare autosomal dominant form of dyshidrotic eczema have identified a locus on chromosome 18q22.1–18q22.3 that may contain the responsible gene(s) [35]. There are 17 genes within this locus, some of which are calcium-dependent adhesion molecules, such as cadherins, which are important in maintaining the structural integrity of epithelial tissues including the skin .
1.3 Nummular Eczema
1.3.1 Clinical Features
The specific etiology of nummular eczema is unknown in children, but associations include xerosis and exposure to various contact allergens in adult patients [36]. It occurs with increased frequency in children with atopic dermatitis but can be seen independently as well [36]. Nummular eczema is characterized by well-circumscribed, erythematous, scaled to exudative pruritic plaques (Fig. 1.7). It typically presents with a relapsing course, although it may abate after puberty.
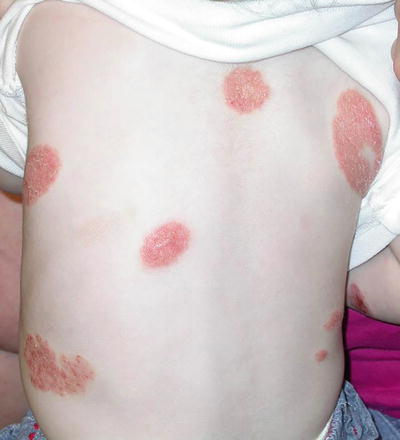
Fig. 1.7
Nummular eczema presents as erythematous scaly circular plaques on the back of an infant
1.3.2 Histology
Nummular eczema demonstrates the classic histologic features of spongiotic dermatitis with no specific distinguishing characteristics (Fig. 1.8). Acute lesions demonstrate marked spongiosis, Langerhans cell abscesses within the epidermis and a brisk lymphocytic infiltrate around the superficial vascular plexus, often with associated eosinophils [37]. Fully developed lesions contain parakeratosis and serum within the stratum corneum, slightly less intraepidermal spongiosis and fewer Langherans cell microabscesses (Fig. 1.9). The dermal inflammatory infiltrate is less intense in chronic lesions than in acute lesions. Well-established chronic lesions are indistinguishable from lichen simplex chronicus .
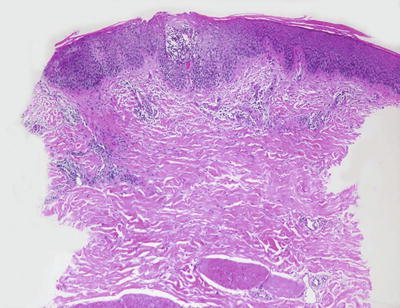
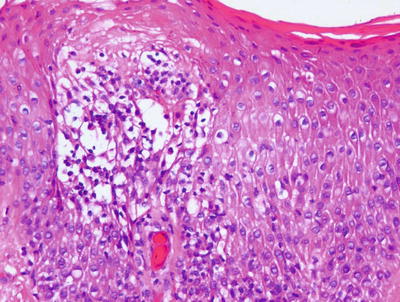
Fig. 1.8
Nummular eczema is a spongiotic dermatitis that displays only a mild superficial perivascular lymphocytic inflammatory infiltrate
Fig. 1.9
Parakeratosis, spongiosis, and microvesiculation are present in most cases of nummular eczema
1.3.3 Pathogenesis
A number of factors have been implicated in the pathogenesis of nummular eczema, including the state of hydration of the skin, infections by house dust mites and Candida albicans , isotretinoin and gold therapy, mercury in dental amalgams, and internal infections of the teeth and respiratory tract [36]. Nummular eczema is likely to be caused by a combination of skin lipid barrier dysfunction and dysregulated immunologic response. Hypersensitivity to metals in the mouth may drive an immune response that results in nummular eczema. Drugs, such as interferon, ribavirin, and inhibitors of tumor necrosis factor (TNF) , can induce generalized nummular lesions [38, 39]. Nummular eczema is pruritic, and mast cells, which are increased in number in this disorder, play a role in pruritus [40–42]. Contributing molecular factors to pruritus include the release of histamine and other inflammatory mediators from mast cells that interact with nerve fibers, and the presence of substance P and calcitonin gene-related peptide, which are increased in eczema skin [41].
1.4 Contact Dermatitis
1.4.1 Clinical Features
Allergic contact dermatitis is a delayed type IV hypersensitivity reaction with apparent increased occurrence in children with incidence rates of 14–70 %, mimicking or exceeding those of adults [43]. Patch testing is the gold standard for diagnosis of allergic contact dermatitis with the top ten allergens in children being nickel (metal in jewelry, belt buckles, buttons), neomycin (topical antibiotic), balsam of peru/fragrance mix (fragrance), thimerosal (preservative), cobalt (metal), chromate (metal in tanned leathers, paints), thiuram (rubber accelerant in elastic bands, shoes, gloves), lanolin (wool wax alcohol found in emollients), formaldehyde (preservative in cosmetic and hygiene products), and paraphenylenediamine (oxidative chemical in hair dye, black henna tattoos) [43].
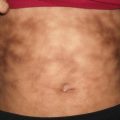
Disease presentation is characterized by pruritic, erythematous patches and plaques localized to the site of contact with the suspected allergen (Figs. 1.10 and 1.11). In more acute variants, erythematous, edematous plaques are present that may or may not vesiculate with secondary serous drainage. In contrast, chronic allergic contact dermatitis is typified by a more insidious onset of lichenified plaques or a generalized eczematous response (namely, auto-sensitization or id reaction). The prognosis is good with resolution of dermatitis if the offending agent is avoided.
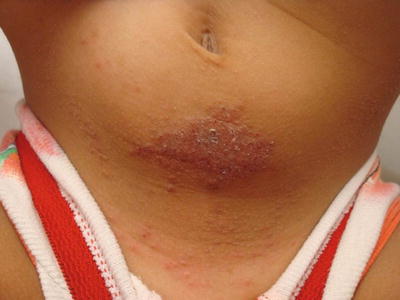
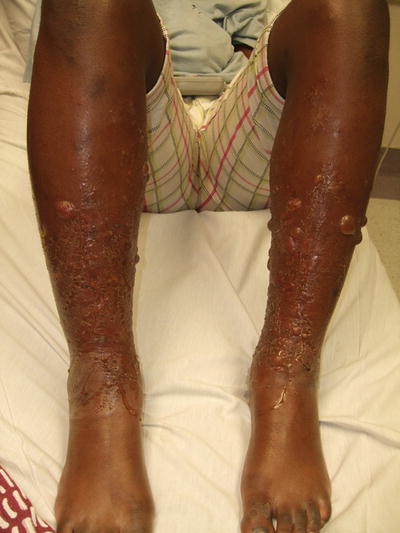
Fig. 1.10
Allergic contact dermatitis presents as an erythematous scaly plaque on the lower abdomen of a toddler due to contact with nickel
Fig. 1.11
Allergic contact dermatitis shows diffuse erythema, edema, vesicles, and bullae on the legs of a child
Irritant contact dermatitis is the most common subtype of contact dermatitis [44]. It requires no prior sensitization and can be seen in any person exposed to the offending agent given the appropriate clinical scenario. Patients present with eczematous dermatitis at the point of contact, with sparing at areas where the skin is protected or not exposed (Fig. 1.12). Resolution with or without secondary dyschromia is seen once the eliciting agent is eliminated.

Fig. 1.12
Irritant contact dermatitis presents as erythematous patches over skin surfaces in the diaper area due to fecal soilage
1.4.2 Histology
Contact dermatitis can be divided into allergic contact dermatitis and irritant contact dermatitis and there are subtle histologic differences that may allow for distinction on biopsy.
Allergic contact dermatitis is a spongiotic process, characterized by parakeratosis (except in the acute state), marked intraepidermal edema and abundant Langerhans cell microabscesses [45] (Fig. 1.13). In florid cases, the spongiosis results in microvesiculation. Eosinophils are abundant within the dermis and may extend into the epidermis (Fig. 1.14). A brisk lymphohistiocytic inflammatory infiltrate is present surrounding vessels of the superficial vascular plexus. Ordinarily, the inflammation does not extend into the lower portions of the dermis [46, 47].
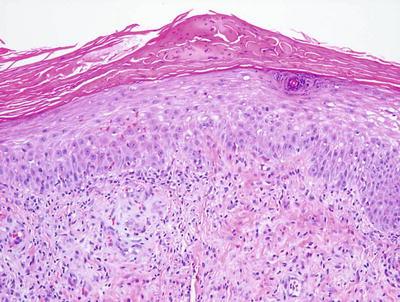
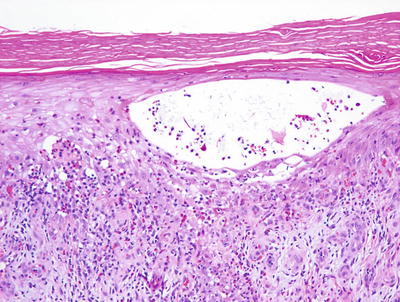
Fig. 1.13
A serum and neutrophil crust overlies a spongiotic epidermis in allergic contact dermatitis
Fig. 1.14
Allergic contact dermatitis frequently displays spongiotic microvesicles that contain eosinophils. Abundant eosinophils are characteristic of this disease
Irritant contact dermatitis has similar changes; however, eosinophils are much less common and may be absent in many cases. Occasional dying keratinocytes may be present as a result of direct cytotoxicity in some cases. As a result of severe toxicity, extensive epidermal necrosis may be present [47, 48].
In chronic stages, both allergic and contact dermatitis demonstrate hyperkeratosis with less parakeratosis, less prominent spongiosis, less intense inflammatory infiltrates and elongation of rete ridges, which are changes reminiscent of lichen simplex chronicus .
1.4.3 Pathogenesis
Allergic contact dermatitis is a T-lymphocyte mediated, delayed-type hypersensitivity reaction elicited by the contact of the skin with a chemical in an individual who has been previously sensitized to the same chemical [49]. The initial sensitization phase involves innate immune response to skin contact with small chemicals. These chemicals are haptens, which are low molecular weight molecules that can penetrate the skin. They can be naturally occurring substances, such as urushiol found in poison ivy, synthetic compounds, dyes, fragrances, drugs, and metal cations (such as nickel and chromium which are common allergic contact dermatitis-associated haptens) [50]. By themselves, haptens are not immunogenic, but when bound to a protein carrier, they can be recognized by the immune system.
The sensitization phase in allergic contact dermatitis requires skin Langerhans cells and dendritic cells that act as antigen presenting cells to present contact allergens to naïve T lymphocytes in the draining lymph nodes [51]. Dendritic cells in the skin interact with keratinocytes, migrate to the local draining lymph nodes and act as antigen-presenting cells to prime naive T lymphocytes. This results in the priming of CD8+ cytotoxic T lymphocytes and type 1 T-helper lymphocytes (Th1) to generate chemical-specific, skin-homing T cell repertoire [52]. There is increased number of allergen-specific circulating T lymphocytes in the blood of patients with allergic contact dermatitis [53–55]. Upon re-exposure to the same chemical, CD8+ T lymphocytes home in and infiltrate the skin, inducing keratinocyte apoptosis via the perforin/granzyme and Fas/Fas ligand pathways [56, 57]. CD4+ CD25+ regulatory T cells (Tregs), which regulate antigen sensitization process, are also important. Allergic contact dermatitis has been found to be associated with defects in Treg cells [58–60].
Small chemicals act as haptens to induce the release of damage-associated molecular pattern (DAMP) molecules and toll-like receptor (TLR) stimulation with the release of pro-inflammatory cytokines, such as interleukin-1β and tumor necrosis factor-α, as well as the recruitment Th1 and IL-17-producing T helper cells (Th17) [53, 54]. TLR2 and TLR4 in particular are important in ACD, and they are activated by DAMP molecules. TLR2 and TLR4 activation leads to the production of interleukins, IFN-γ and TNF-α, which are potent activators of keratinocytes and promote the recruitment of effector T lymphocytes and the inflammatory response [61, 62].
1.5 Autosensitization Dermatitis/Id Reaction
1.5.1 Clinical Features
Auto-sensitization dermatitis or “id reaction” is a cutaneous hypersensitivity mediated by host T-cell lymphocytes to a number of stimuli including infectious insults and primary inflammatory skin conditions [63]. Disease onset may follow initiation of antimicrobial therapy or may occur subsequent to flare of the responsible dermatitis.
Patients typically present with the acute onset of generalized, symmetric, pruritic, edematous papules and papulovesicles. Clinical features vary and are dependent upon the inciting agent and the secondary host response [63]. Diagnosis is suggested by resolution of acute eruption following appropriate treatment of the primary inciting event (Figs. 1.15 and 1.16). The prognosis is good but recurrence is common if the primary source is not appropriately eliminated.
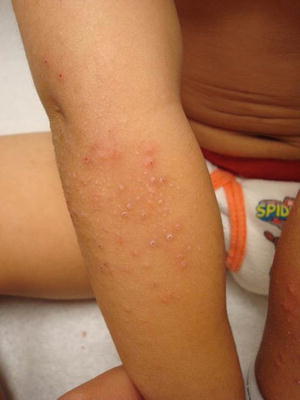
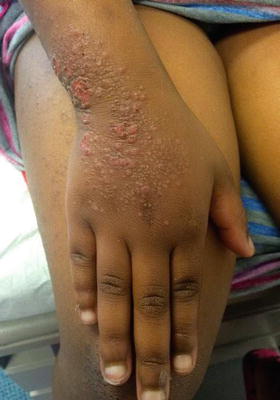
Fig. 1.15
Autosensitization/Id reaction presents as monomorphous scaly papules over the extensor arm
Fig. 1.16
Autosensitization/Id reaction may present as monomorphous scaly papules coalescing into a plaque as seen here over the dorsal hand and wrist of a child
1.5.2 Histology
Histologic features of autosensitization dermatitis include parakeratosis admixed with serum in the stratum corneum, intraepidermal spongiosis with occasional microvesiculation, and a lymphohistiocytic infiltrate surrounding vessels within the superficial vascular plexus (Figs. 1.17, 1.18 and 1.19). Eosinophils are generally not abundant, in contrast to allergic contact dermatitis. The deep dermis is usually not involved [63, 64]. As lesions become more chronic, there is a tendency for diminution of spongiosis and inflammation, and increased acanthosis of the epidermis.
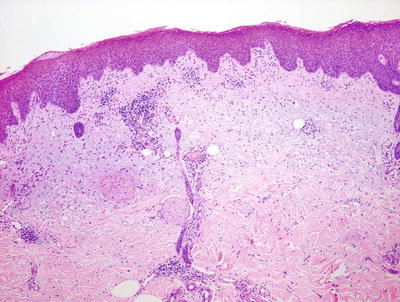
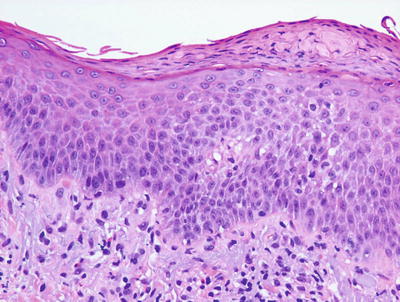
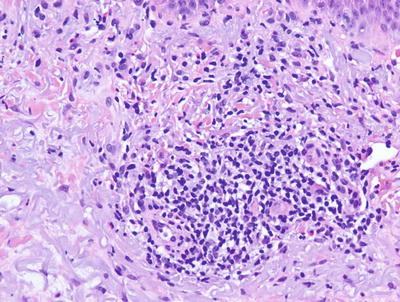
Fig. 1.17
An id reaction is characterized by a mild spongiotic dermatitis and a superficial perivascular lymphocytic inflammatory infiltrate
Fig. 1.18
Parakeratosis is present overlying a spongiotic epidermis in an id reaction
Fig. 1.19
An id reaction demonstrates a perivascular lymphocytic infiltrate with scattered eosinophils
1.5.3 Pathogenesis
Autosensitization (id reaction) is a delayed hypersensitivity reaction to prior antigen exposure [65]. It is a secondary immunologic response to circulating antibodies and activated T lymphocytes directed against a sensitizing antigen. For example, an id reaction to fungal organisms may be due to a chain reaction induced by fungal antigens at the site of infection, opsonization of antigens by host antibodies, development of memory T lymphocytes and dissemination of these cells systemically [66]. Id reaction can present as localized vesicular lesions in tinea pedis, erythema nodosum in bacterial and deep fungal infections, and erythema multiforme in herpetic infections [63].
1.6 Photoallergic Dermatitis
1.6.1 Clinical Features
Photoallergic dermatitis is thought to represent a cell-mediated delayed hypersensitivity reaction, requiring prior sensitization to an exogenous photoallergen and subsequent ultraviolet light (UV) radiation, namely UVA [67]. Initial sensitization and development of skin changes may occur 1–2 days after exposure to the allergen and sunlight [67]. On subsequent exposures, cutaneous reaction typically occurs within 24 hours even with minimal sun exposure.
Urticarial and eczematous lesions have been described with the physical exam mimicking that of allergic contact dermatitis, although skin changes are most obvious in areas that are more heavily exposed to the sun. Secondary post-inflammatory hyperpigmentation is rare [67]. The prognosis is good when the causative agent is avoided and photoprotection is employed.
1.6.2 Histology
Photoallergic dermatitis is histologically indistinguishable from acute contact dermatitis. Histologic sections demonstrate a stratum corneum that may be unremarkable in the acute state, or contain parakeratotic nuclei and serum in the subacute phase. The epidermis is markedly spongiotic with occasional microvesicles [68, 69] (Fig. 1.20). Clusters of Langerhans cells and scattered intraepidermal eosinophils and lymphocytes are present. Dying keratinocytes are not prominent. Within the papillary dermis, there is edema and a moderately intense collection of lymphocytes and eosinophils surrounding the superficial vascular plexus (Fig. 1.21). In late stages, the epidermis becomes more acanthotic and spongiosis dissipates .
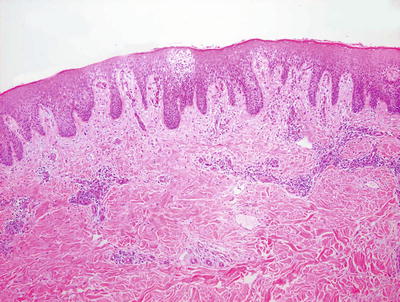
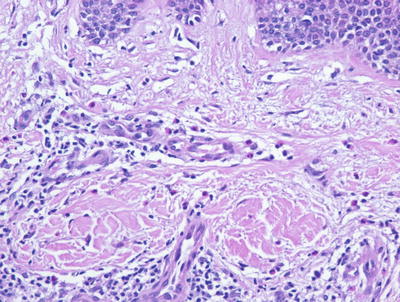
Fig. 1.20
Photoallergic dermatitis is a spongiotic dermatitis that results in intraepidermal edema, exocytosis of lymphocytes into the epidermis, and microvesiculation when severe
Fig. 1.21
The inflammatory infiltrate in photoallergic dermatitis includes lymphocytes and abundant eosinophils within the dermis and sometimes extending into the epidermis
1.6.3 Pathogenesis
Photoallergic dermatitis to allergens, such as plant materials (phytophotodermatitis), results from skin exposure to sap from a plant that reacts with long wavelength ultraviolet light (UV) [70]. Photoallergens may form from haptens that become covalently coupled to a carrier protein via UV irradiation [71]. UVA, but not UVB, is the UV spectrum of this photoreactivity. Cells that are modified by photoallergens are capable of inducing the proliferation of sensitized T lymphocytes and eliciting an immune response [72, 73]. In addition to T lymphocytes, skin Langerhans cells are important in the pathogenesis of photodermatitis and function as antigen-presenting cells of photoallergens to sensitized T lymphocytes [71].
1.7 Pityriasis Alba
1.7.1 Clinical Features
Pityriasis alba most commonly affects children between 3 and 16 years of age with an incidence of 2 to 5 % in pediatric populations [74]. Prevalence is higher in darker-skin individuals. No gender predilection exists.
Pityriasis alba is characterized by hypopigmented patches with subtle scaling, most commonly on the face but also occurring on the neck, upper trunk, and proximal extremities (Fig. 1.22). Skin changes typically are most prominent after sun exposure, as affected areas do not darken with ultraviolet light exposure. Although pityriasis alba is a chronic condition with a relapsing course, it has a good prognosis with eventual spontaneous resolution.
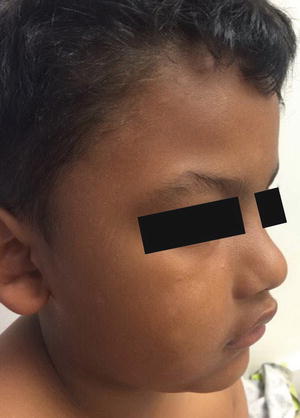
Fig. 1.22
Pityrisis alba shows scattered, poorly defined hypopigmented macules and patches over the cheeks
1.7.2 Histology
Histologic changes in pityriasis alba are nonspecific. They include hyperkeratosis and parakeratosis, mild spongiosis, and plugging of follicles in some cases [75]. Atrophic sebaceous glands have been described in some patients [76]. There is a superficial perivascular lymphocytic infiltrate with focal exocytosis. The most specific changes are those of decreased numbers of melanocytes in the epidermis [77]. However, use of melanocyte-specific markers suggest that there is no significant decrease in numbers of melanocytes, but there is diminished melanin production [78]. Residual melanocytes within affected areas demonstrate decreased numbers of melanosomes on ultrastructural analysis [77, 78].
1.8 Gianotti–Crosti Syndrome
1.8.1 Clinical Features
Gianotti–Crosti syndrome is a dermatitis almost exclusively seen in children, with peak incidence between 1 and 6 years of age (range of 3 months old to 15 years old) [79]. It is thought to be a cutaneous response to a viral insult and may be seen following vaccinations, although reports of similar eruptions following a bacterial infection have been reported [79–81].
The syndrome is characterized by multiple symmetric, monomorphic and erythematous (pink–reddish brown) 1–5 mm papules or papulovesicles on the face, buttocks, and extremities (Figs. 1.23, 1.24, and 1.25). Distinct lesions may coalesce in some areas. A preceding prodrome may be present and includes malaise, fever, diarrhea, and concomitant lymphadenopathy [79]. Skin lesions resolve spontaneously within weeks to months without secondary dyschromia or scarring. Hepatitis and other visceral involvement are rare with no reports of long-term sequelae or morbidity [79].
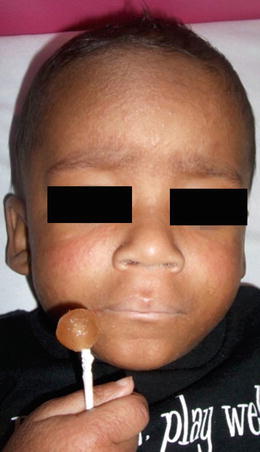
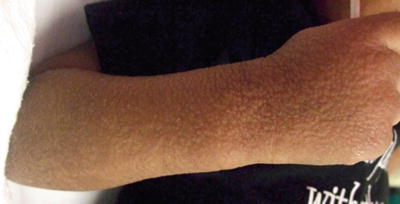
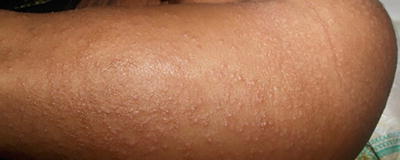
Fig. 1.23
Diffuse small monomorphic papules on the face of a child with Gianotti–Crosti syndrome
Fig. 1.24
Diffuse small papules present on the extensor arm and hand in the same child with Gianotti–Crosti syndrome
Fig. 1.25
This same child with Gianotti–Crosti syndrome had similar diffuse monomorphic papules on the legs and buttocks
1.8.2 Histology
Gianotti–Crosti syndrome is characterized by a mild spongiotic process, often with pronounced papillary dermal edema (Fig. 1.26). Focal parakeratosis is variably present. A superficial perivascular lymphohistiocytic infiltrate demonstrates only rare eosinophils. In rare cases, spongiotic microvesicles containing Langerhans cells may be present adjacent to areas of epidermal acanthosis. Some authors have suggested that papillary dermal edema is most prominent in cases associated with Epstein–Barr virus infection [82]. Extravasated erythrocytes have been described in some cases. In rare cases, an interface dermatitis with scattered basal vacuolization has been described [83].
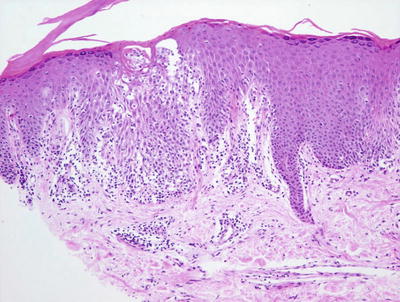
Fig. 1.26
Gianotti–Crosti disease demonstrates nonspecific spongiotic dermatitis with a lymphocytic infiltrate and scattered exocytosis
1.8.3 Pathogenesis
Viral infection is an important contributing factor in the pathogenesis of Gianotti–Crosti Syndrome. Epstein–Barr virus (EBV) is a common viral infection associated with the disease [84, 85]. Viral antigens and antiviral antibodies have been found in the serum of patients. Hepatitis B virus (HBV) is another common viral infection associated with Gianotti–Crosti syndrome, although there has been a decline in HBV-associated disease due to anti-HBV immunizations [86, 87]. Atopic dermatitis has also been implicated in disease pathogenesis. In a case–control study, children with Gianotti–Crosti syndrome were more likely to have atopic dermatitis or a family history of atopy than children without disease, who were being evaluated for recurrent infections (24.1 % versus 6.8 % for atopic dermatitis, and 51.7 % versus 31 % for family history of atopy) [88]. Gianotti–Crosti syndrome commonly occurs in children, and the disease is less severe in children than in adults. A possible explanation for the higher frequency of disease in children is that EBV infection is more common in children. The immature immune system of infants and children, and the resulting differences in response to viral infections in children may be a basis for different disease severity in children as compared to adults .
1.9 Intertrigo
1.9.1 Clinical Features
Intertrigo is a frictional dermatitis caused by persistent rubbing of adjacent skin surfaces. The condition is frequently exacerbated by excess moisture within the deep skin folds of the affected areas. Secondary infection may be seen, most commonly with candida fungal species, although reports of infection by group A β-hemolytic streptococcus, Staphylococcus aureus and mixed bacterial species have been documented as well [89].
Intertrigo is characterized by macerated, erythematous patches at the skin folds. Candidal intertrigo may be distinguished by presence of satellite papules and pustules. Bacterial intertrigo, on the other hand, has circumscribed, weeping or moist, “fiery red” erythema, possibly with associated malodor and tenderness (Fig. 1.27). Clearance can be achieved with measures to minimize friction and treatment of any secondary infections.
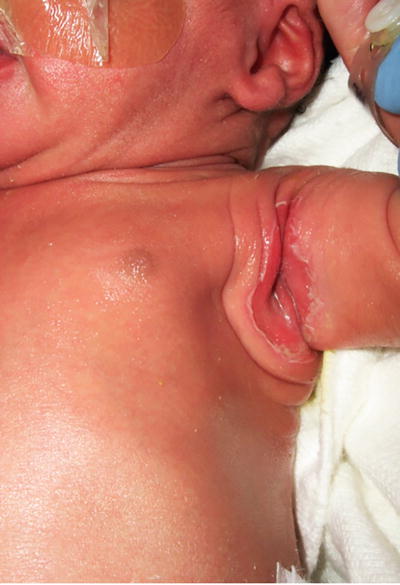
Fig. 1.27
Bacterial intertrigo presents in this infant as an erythematous erosion in the axillary crease
1.9.2 Histology
Intertrigo has no characteristic histologic changes [90]. Mild spongiosis may be present in the epidermis in some cases. In many cases, there is superimposed infection with bacteria and fungi (Fig. 1.28). In those cases, changes suggestive of impetigo or dermatophyte infection may be present.
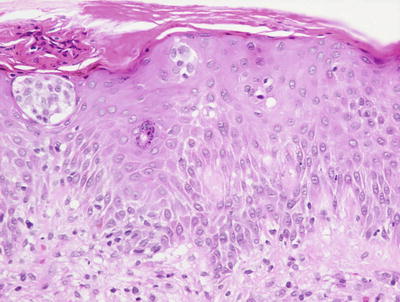
Fig. 1.28
The findings in intertrigo include a nonspecific spongiotic dermatitis, here seen containing Langerhans cell abscess within the epidermis
1.9.3 Pathogenesis
Intertrigo is precipitated by moisture trapped in deep skin folds [90, 91]. Hyper-hydration and friction between skin folds can lead to maceration of the stratum corneum and damages to the epidermis. The condition can progress to severe inflammation and erosion of the skin barrier, thus facilitating the growth of microorganisms, such as Staphylococcus aureus , group A β-hemolytic streptococcus, and fungi.
Candida albicans is a fungus that has predilection to colonize macerated folds of skin and is often associated with intertrigo. Candida infection and intertrigo occur more frequently in patients with diabetes mellitus because these patients have a higher pH in intertriginous areas, which may be a factor that promotes the susceptibility of these patients to Candida infection [92, 93]. Other dermatophytes, namely Trichophyton rubrum, Trichophyton mentagrophytes and Epidermophyton floccosum, are often found in interdigital intertrigo in the hands and feet [90].
1.10 Periorificial Dermatitis
1.10.1 Clinical Features
Periorificial dermatitis is an eczematous and acneiform eruption of unknown etiology seen across a spectrum of pediatric patients from infants to older adolescents, although most cases are reported in those less than 5 years old [94]. A majority of children with disease have a history of steroid exposure, whether it is topical, inhaled or by systemic administration [95]. Sub-variants of the disorder include granulomatous periorificial dermatitis and facial Afro-Caribbean childhood eruption (FACE), both of which are seen more commonly in persons with darker skin. Isolated perioral lesions are more commonly seen, although lesions can be more generalized with skin changes around the nose, eyes, and mouth and even at the genital orifices [95].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


