(10.1)
10.2 Basis for Prodrugs as Penetration Enhancers
Although increasing the “push” can be easily accomplished by manipulating the components of the vehicle in which the drug is applied (its formulation), increasing the “pull” can be more easily accomplished using a prodrug approach that changes the solubility properties of the drug. A prodrug is a chemically or enzymatically reversible derivative of a parent drug that improves the physicochemical or biological properties of the parent drug molecule to overcome some intrinsic problem associated with its therapeutic use: in this case, poor solubility in the skin and hence low topical delivery (Sloan 1992). The particular combination of functional groups that is added to the parent drug is called the promoiety, and the reversible connection between the promoiety and the parent drug is called the enabling functional group. A prodrug approach, then, can be envisaged as a 1:1 molecular combination of the drug and a promoiety that contains functional groups that will increase its solubility in the skin (Sloan and Wasdo 2003). This prodrug approach stands in sharp contrast to most formulation approaches where large molar excesses of penetration enhancers as vehicle components are routinely needed to increase S M1 for the drug.
What are the properties of the functional groups in the promoiety which, when added to the parent drug, could be reasonably expected to cause an increase in S M1 of the resulting prodrug compared to the parent drug and hence to cause an increase in its maximum flux, J M? Since it is difficult to measure S M1 of the prodrug, it is more convenient to measure its J M in diffusion cell experiments and assume, based on Fick’s law Eq. 10.1, that there is a direct relationship between increased J M and increased S M1. Using increases in J M as the criterion for increased S M1, it has been observed for quite some time that for homologous series of more lipophilic prodrugs that the more water soluble members of the series gave the greatest increase in J M and not the more lipid soluble members (Sloan 1989, 1992; Sloan et al. 1984). In order to account for these qualitative observations, S M1 in Fick’s law Eq. 10.1 was expanded mathematically to include dependence on solubility in a lipid, S LIPID, and in water, S AQ. This form of Fick’s law is the Roberts-Sloan (RS) Eq. 10.2 (Roberts and Sloan 1999): a transformation of the popular, but very specific, Potts-Guy (PG) Eq. 10.3 (Potts and Guy 1992) into more general, useful terms.
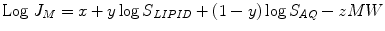
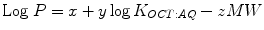
(10.2)
(10.3)
When a database of those homologous series of more lipid soluble prodrugs (n = 42) comprised of their molecular weights, MW, their solubilities in isopropyl myristate (IPM), S IPM (S IPM = S LIPID in Eq. 10.2), and in water, S AQ, and their maximum fluxes from IPM through hairless mouse skin, J MMIPM, were collected and fitted to Eq. 10.2, the values for the coefficients were x = −0.211, y = 0.534, z = 0.00364, and r 2 = 0.937 (Roberts and Sloan 1999). The size of the J MMIPM database has since been increased to n = 94, and the values for the coefficients are now x = −0.377, y = 0.527, z = 0.00346, and r 2 = 0.900 (Majumdar et al. 2012). The maximum fluxes of prodrugs and non-prodrug through human skin in vitro and in vivo, respectively, from mineral oil (MO), J MHMO, their solubilities in mineral oil, S MO (S MO = S LIPID in Eq. 10.2), and in water, S AQ, and their MW also gave good fit to Eq. 10.2: x = −1.83, y = 0.462, z = 0.00153, and r 2 = 0.80 for n = 30 prodrugs (Sloan et al. 2011); x = −1.459, y = 0.72, z = 0.00013, and r 2 = 0.934 for n = 10 nonsteriodal anti-inflammatory drugs (Wenkers and Lippold 1999; Roberts and Sloan 2001). Thus, good fits to Eq. 10.2 are obtained if the vehicle is a lipid (IPM or MO) and the lipid solubility of the permeant, S LIPID, and S AQ are independent valuables.
A similar strong dependence of maximum flux through hairless mouse from water, J MMAQ, on S IPM (S IPM = S LIPID in Eq. 10.2) and S AQ for some of the members of the n = 94 J MMIPM database was observed where x = −2.30, y = 0.575, z = 0.0016, and r 2 = 0.903 for n = 32 (Sloan et al. 2003; Wasdo et al. 2009). Also a strong dependence of maximum flux through human skin in vitro from water, J MHAQ, on the solubilities of the permeants in octanol, S OCT (S OCT = S LIPID in Eq. 10.2) and S AQ, was observed where x = −2.506, y = 0.538, z = 0.00402, and r 2 = 0.839 for n = 185 (Juntunen et al. 2008). Even maximum flux through silicone membranes from water, J MPAQ, for some of the members of the n = 94 J MMIPM database was found to be dependent on S IPM (S IPM = S LIPID in Eq. 10.2) and S AQ where x = −1.837, y = 0.742, z = 0.00435, and r 2 = 0.86 for n = 38 (Synovec et al. 2013). Thus, good fits to Eq. 10.2 are obtained regardless of whether the membrane is mouse, human, or silicone and regardless of whether the vehicle is a lipid or aqueous. Since the solubilities of the permeant in a lipid and in water are both necessary to define maximum flux, functional groups should be incorporated into the promoieties of prodrugs that can ideally increase both lipid and aqueous solubilities to increase maximum flux and by inference S M1.
The reason that increasing both lipid and aqueous solubilities of the drug is important to increasing its solubility in skin, and hence its topical delivery, can be found in the structure of the barrier to topical delivery – the intercellular compartment of the stratum corneum (SC). The intercellular compartment consists of lamellar double bilayers comprised of lipid components such as ceramides, cholesterol, and fatty acids which have polar groups attached to them. These polar head groups have water associated with them so that for a permeant to cross these bilayers perpendicular to the axis of the bilayers, it must alternately cross lipid and aqueous phases (Sloan and Wasdo 2003; Sloan et al. 1984, 2011a, b). Thus, a balance of solubility in both lipid and aqueous phases by the drug (or increased lipid and aqueous solubility by its prodrug) is necessary for its most efficient permeation of the intercellular compartment of the SC. The agreement between the experimentally measurable physicochemical parameters in the theoretically derived Roberts-Sloan equation and in the biochemically based biphasic solubility model (Sloan et al. 2011a, b) for the barrier to permeation is encouraging.
10.3 Acyl Versus Soft Alkyl Promoieties
The promoieties that have been used to increase lipid and aqueous solubilities can be divided into two types based on whether they are attached directly to the functional group in the parent drug that is to be modified or indirectly through a methylene or vinylogous methylene (aryl methylene) spacer (Sloan 1989, 1992; Sloan and Wasdo 2003). In each type, the enabling functional group is usually a carbonyl-type functional group because of its sensitivity to cleavage by chemical or enzymatic hydrolysis. Generally these types have been referred as acyl and soft alkyl-type promoieties, respectively. Cleavage of the acyl-type promoiety regenerates the parent drug directly while cleavage of the acyl group in the soft alkyl promoiety generates an intermediate drug–X–CHR–X′H from drug–X–CHR–X′–(C = X″)–X′′′R′: X, X′, X″, and X′′′ can be O, N, or S and R and R′ can be alkyl or aryl. The intermediate is designed to be intrinsically unstable and undergo rapid and complete chemical hydrolysis to the parent drug–X–H. The advantage of the soft alkyl prodrug approach is that the stability of the prodrug (as well as its attendant physicochemical properties) is not limited by the functional group in the parent drug to which it is attached. Generally, changing X will change the biochemical and/or pharmacological activity of the drug, but changing X′ to obtain a more or less stable or more or less soluble prodrug will not. Of course X″ and X′′′ can be changed in the same ways that they could have been if an acyl prodrug approach had been used.
10.4 Mechanisms for Penetration Enhancement
10.4.1 Decrease Crystal Lattice Energy by Masking Hydrogen Bond Donor Functional Groups
Regardless of whether the prodrug is derived from an acyl or soft alkyl-type promoiety, there are two general mechanisms by which both types of promoieties can increase both lipid and aqueous solubilities. The first mechanism has its basis in decreasing the crystal lattice energy of the parent drug by modifying polar groups capable of forming intermolecular hydrogen bonds. In many if not most drug molecules, the X in drug–X–H is a heteroatom which causes X–H to be polarized because of the difference in electronegativities between X and H. This polarized drug–X–H bond is capable of forming intermolecular hydrogen bonds within the crystal lattice which leads to low solubilities especially in lipids but also frequently in water. The polarization is further attenuated if an electron withdrawing carbonyl-type functional group is attached to X–H to give drug–(O = C)–X–H. Examples of this type of drug molecule, which can be measurably but not highly ionized at physiological pH, include heterocycles such as 5-flurouracil (5-FU) (drug–(O = C)–NH) and 6-mercaptopurine (6-MP) (drug–(S = C)–NH) which are very high melting and exhibit low solubilities in both water and lipids. In other examples such as parent drugs containing a carboxylic acid functional group (drug–(O = C) –OH), the functional group is so highly polarized that it becomes highly ionized at physiological pH which does not allow it to readily cross the lipid phase of the alternating lipid-aqueous phases of the biological barrier. An important class of drugs that belong to this category is the nonsteroidal anti-inflammatory drugs. Another example of this class are the nucleotide-based drugs where the highly ionized functional group is a phosphate group. Simply masking the hydrogen bond donating abilities of the functional group by replacing the H in the drug–X–H with either an acyl or soft alkyl group decreases the melting point (mp) and increases the lipid solubility (S LIPID) as well as frequently increasing the aqueous solubility (S AQ) of the prodrug compared to the parent drug, especially for the shorter alkyl chain members of a homologous series (Sloan 1989).
Examples of the results that can be obtained by masking the polar functional groups in drugs to increase S LIPID (S IPM) and S AQ and to increase topical delivery of the parent drug are several prodrugs of 5-FU.
Table 10.1
Prodrugs of 5-fluorouracil
Prodrugs, R = a | mpb | S IPM c | S AQ c, d | Log K IPM: AQ e | J MMIPM f |
|---|---|---|---|---|---|
1–AAC–5–FU | |||||
1, C1NHC = O | 212 | 0.30 | 3.69 | −1.09 | 0.208 |
2, C2NHC = O | 180 | 2.79 | 7.76 | −0.44 | 0.600 |
3, C3NHC = O | 139 | 12.4 | 8.98 | 0.14 | 0.746 |
4, C4NHC = O | 133 | 24.6 | 5.11 | 0.68 | 0.515 |
5, C6NHC = O | 113 | 44.9 | 0.36 | 2.09 | – |
6, C8NHC = O | 91 | 46.9 | 0.030 | 3.21 | 0.060 |
1-AOC-5-FU | |||||
7, C1OC = O | 160 | 2.13 | 112 | −1.72 | 2.62 |
8, C2OC = O | 128 | 13.1 | 175 | −1.12 | 5.92 |
9, C3OC = O | 126 | 15.2 | 42.2 | −0.44 | 2.31 |
10, C4OC = O | 98 | 33.8 | 24.1 | 0.15 | 2.23 |
11, C6OC = O | 67 | 153 | 4.94 | 1.49 | 1.54 |
12, C8OC = O | 98 | 36.2 | 0.13 | 2.45 | 0.29 |
1-AC-5-FU | |||||
13, C1C = O | 130 | 22.1 | 120 | −0.73 | 9.3 |
14, C2C = O | 131 | 36.4 | 47.6 | −0.12 | 4.3 |
15, C3C = O | 146 | 17.4 | 6.50 | 0.43 | 1.3 |
16, C4C = O | 121 | 39.2 | 3.48 | 1.05 | 1.0 |
17, C5C = O | 102 | 112.7 | 2.94 | 1.58 | 1.1 |
18, C7C = O | 84 | 110.7 | 0.15 | 2.88 | 0.60 |
1-ACOM-5-FU | |||||
19, C1(C = O)OCH2 | 124 | 3.29 | 183 | −1.74 | 2.88 |
20, C2(C = O)OCH2 | 102 | 9.83 | 167 | −1.23 | 3.82 |
21, C3(C = O)OCH2 | 91 | 14.4 | 42.4 | −0.47 | 2.57 |
22, C4(C = O)OCH2 | 88 | 14.8 | 12.3 | 0.08 | 1.29 |
23, C5(C = O)OCH2 | 91 | 14.7 | 2.23 | 0.82 | 0.56 |
24, C7(C = O)OCH2 | 108 | 9.99 | 0.17 | 1.77 | 0.12 |
5-FU, H | 284 | 0.049 | 85.4g | −3.24h | 0.240 |
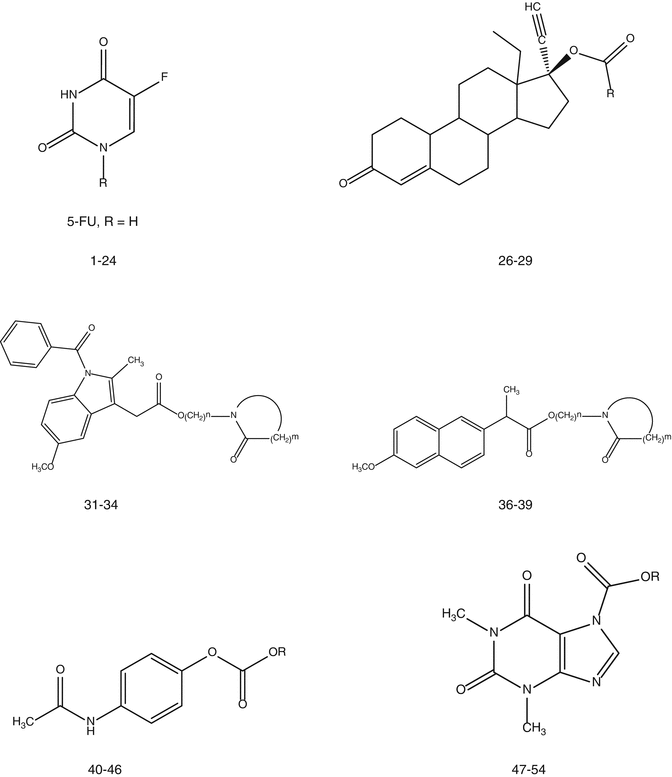
The mp, S AQ, S IPM, log partition coefficients between IPM and pH 4.0 buffer (log K IPM:AQ), and rates of delivery of total 5-FU containing species through hairless mouse skin from an IPM vehicle in vitro (J MMIPM) for four different series of prodrug of 5-FU are given in Table 10.1: three acyl types and a one soft alkyl type. The first acyl type of prodrug of 5-FU that was evaluated for its ability to increase the delivery of 5-FU was the alkylaminocarbonyl-5-FU (1-AAC-5-FU) prodrugs (Table 10.1 and Fig. 10.1). Initially only the longer alkyl chain members of the series were evaluated (4–6) (Sasaki et al. 1990), but subsequently the shorter alkyl chain members (1–3) were evaluated, and one of them, 3, was found to give the greatest increase in the delivery of the total 5-FU containing species, J MMIPM (Sloan et al. 1993). All of the 1-AAC-5-FU prodrugs exhibited lower mp than 5-FU and all of them were more soluble in IPM than 5-FU: from 6 times for 1 to almost 1,000 times for 6. However, the most lipid soluble member evaluated, 6, gave only 0.25 times the flux of 5-FU. None of the 1-AAC-5-FU prodrugs was even as soluble in water as 5-FU, and the C3 member (3), not the shortest alkyl chain member of the series (1), gave the highest S AQ value: only 0.11 times S AQ for 5-FU. The C3 member also gave the greatest increase in J MMIPM values for the series, albeit only three times. Thus, as predicted (Sloan 1992, 1989; Sloan and Wasdo 2003; Sloan et al. 1984), for a more lipid soluble homologous series of prodrugs, the more water soluble member gave the highest J MMIPM value. The low increase in J MMIPM can be attributed to the low S AQ values exhibited by the 1-ACC-5-FU prodrugs compared to subsequent series, and the low S AQ values can be attributed to the fact that one of the hydrogen bond donor functional groups, (O = C)–NH, in 5-FU was merely replaced with another hydrogen bond donor group, N–(O = C) –NH, in the promoiety. The potential for forming intermolecular hydrogen bonds was not decreased significantly and the added alkyl group in the promoiety further depressed S AQ
.
Fig. 10.1
Chemical structures of prodrugs for compounds 1-54
The second acyl type of prodrug of 5-FU that was evaluated was the alkyloxycarbonyl-5-FU (1-AOC-5-FU) prodrugs (Table 10.1, Fig. 10.1) (Beall et al. 1994). In this series the hydrogen bond donating group in the parent drug has not been replaced with another hydrogen bond donating group in the promoiety so the mp are somewhat lower than the corresponding members in the 1-AAC-5-FU series except for the C8 member of the series. Consequently, the members of the 1-AOC-5-FU series were also somewhat more soluble in IPM than the members of the 1-AAC-5-FU series except for the C8 member, 12; and the worst member of the series in terms of increased S IPM was 43 times instead of 6 times more soluble in IPM than 5-FU. However, the big difference between the two series was in the S AQ values. Not only were two members of the series more water soluble than 5-FU, 7 and 8 (1.3 and 2 times, respectively), but they were all more water soluble than the corresponding members of the 1-AAC5-FU series (from 30 to 4.3 times). Thus, since the 1-AOC-5-FU series was more soluble in lipids and in water, as predicted (Sloan 1989, 1992; Sloan et al. 1984), they delivered more total 5-FU species through hairless mouse skin than the 1-AAC-5-FU series (from 3 to 12.5 times). Also, as predicted (Sloan 1989, 1992; Sloan et al. 1984) the C2 member, 8, which was the most water soluble member of the series gave the greatest increase in J MMIPM compared to 5-FU (24.7 times), and not the most lipid soluble member of the series, 11. The next most water soluble member, 7, gave the next greatest increase in J MMIPM compared to 5-FU (11 times).
Related posts:
 The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
 Epidermal Lipids and the Intercellular Pathway
Epidermal Lipids and the Intercellular Pathway
 Skin Deep: The Basics of Human Skin Structure and Drug Penetration
Skin Deep: The Basics of Human Skin Structure and Drug Penetration
 Liposomal Gels in Enhancing Skin Delivery of Drugs
Liposomal Gels in Enhancing Skin Delivery of Drugs
 Pickering Emulsions for Controlled Drug Delivery to the Skin
Pickering Emulsions for Controlled Drug Delivery to the Skin
 Formulation of Drug-Cyclodextrin Complexes
Formulation of Drug-Cyclodextrin Complexes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


