Fig. 15.1
Schematic of the human retina. The figure shows the laminated retinal structure with the various cell types arranged in distinct layers
In humans and nonhuman primates, the central region of the retina is enriched with cones and is called the macula. The central part of the macula called fovea centralis has a pit due to the presence of only cone photoreceptors. This is the region of highest density of cone photoreceptors in the eye. The photoreceptors transmit the light information to a group of retinal interneurons called bipolar cells. These cells, in turn, transmit the signals to ganglion cells, either directly or through another class of inhibitory retinal interneurons, called amacrine cells. The ganglion cells are the main output neurons of the retina, whose axons combine to form the optic nerve to send the visual information to central visual centers. Following retinal differentiation, the final cell that comes out of the retinal stem cells is the Müller glial cells.
Outer to the photoreceptor layer is the layer of pigmented cells, called RPE. RPE cells are vital for the maintenance of the rods and cones. The apical end of RPE cells faces the photoreceptors and has microvilli, which completely engulf the outer segments of the photoreceptors. RPE cells perform multiple supportive functions. The RPE layer transports ions, water, and metabolic by-products from the photoreceptor side to the choriocapillaris on the other side. The rod and cone photoreceptors shed approximately 10 % of their outer segments a day. This generates enormous amount of cellular debris into the subretinal space and the RPE cells are responsible for getting rid of photoreceptor disc debris. Additionally, RPE cells are critical for the normal visual cycle. The cells recycle visual cycle component, all-trans retinol, back to 11-cis form. The RPE layer is also part of the blood–retinal barrier, which may play a critical role in immune protection and immune privilege. Finally, the pigmented cells absorb and prevent light scattering thereby aiding in visual acuity.
15.2 Retinal Degenerations
The retina is subject to a number of inherited and acquired forms of retinal degenerative conditions. Age-related macular degeneration (AMD) is a disease of the retina characterized by the degeneration of cells in the RPE and the photoreceptors in the central region of the retina. AMD is one of the leading causes of blindness in the USA. It affects approximately 10 % of the population over 65 and up to 1 in 3 over the age of 80 years. Glaucoma and diabetic retinopathy also lead to retinal neuronal loss. Glaucoma primarily leads to apoptosis of ganglion cells, while diabetic retinopathy leads to loss of most retinal neurons secondary to a vasculopathy. The incidence of both of these diseases increases with age. There are also a number of inherited retinal disorders including Usher’s syndrome and retinitis pigmentosa (RP), which do not manifest until much later in life.
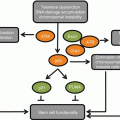
Age-related macular degeneration, the leading cause of worldwide blindness in the elderly, affects approximately 15 million people in the USA alone. About 1.75 million people in USA currently have advanced age-related macular degeneration with associated vision loss, and the number is expected to grow to almost three million by 2020. The disease poses a significant financial burden on the society accounting for $68 billion in direct costs in the USA. The pathogenesis of AMD is unclear and multiple factors may be involved in the progression of the disorder including chronic oxidative stress, lifelong light exposure, small RNA processing defects in the RPE, choriocapillaris atrophy, accumulation of lipofuscin, and inflammation and complement activation (Fig. 15.2) (Jarrett and Boulton 2012; Kaneko et al. 2011; Khan et al. 2006). The disease starts in the macula, the region of central retina with highest visual acuity. There are two main forms of the AMD disorder: dry and wet form. The disease is characterized by the development of drusen which are pathological extracellular deposits containing glycolipids, proteins, and cellular debris, between the RPE and the overlying Bruch’s membrane. Early AMD is associated with more numerous soft drusen in the macula, pigmentary changes in the RPE, and thickening of Bruch’s membrane. Advanced forms of AMD can manifest as either geographic atrophy, which is the dry form, or neovascular/wet form of AMD. Geographic atrophy results in dysfunction and loss of the RPE and degeneration of the underlying photoreceptors occurs. Wet AMD is characterized by the development of choroidal neovascularization following disruption of the Bruch’s membrane, leading to exudation, bleeding, and scar formation.
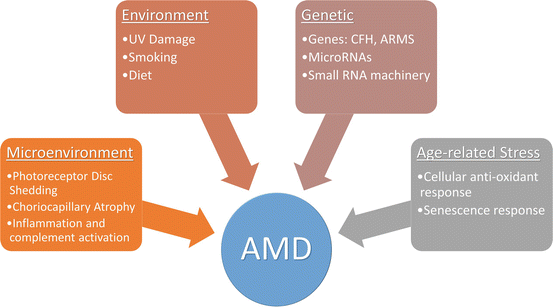
Fig. 15.2
Multifactorial etiology of age-related macular degeneration. A number of genetic, environmental, local microenvironment, and age-associated factors have been linked to the development of the disorder
A number of studies have associated oxidative stress as the key driver to AMD (Jarrett and Boulton 2012). Evidences pointing to this include (a) decrease in microsomal glutathione S-transferase-1, an enzyme that reduces peroxides, oxidized RPE lipids, and oxidized retinoids, (b) increased lipid peroxidation, and (c) age-associated decrease in catalase activity in the human RPE. The resultant oxidant damage manifests in age-related increases in autofluorescent lipofuscin granules especially those containing A2E, a pyridinium bisretinoid by-product of visual pathway component all-trans retinol. A2E is a potent photo-inducible generator of ROS in the RPE (Sparrow and Boulton 2005). Additional cellular changes include 8-oxodG accumulation, lipid peroxidation and glycation of end products, and mitochondrial DNA damage. Linkage and genome-wide association studies have implicated genetic modulators of AMD risk related to many mechanistic pathways, including oxidative stress, complement system dysregulation, DNA repair, mitochondrial dysfunction, neovascularization, and microglial recruitment (Gorin 2012). The Y402H polymorphism in complement factor CFH has been consistently demonstrated as a significant risk factor for AMD (Boon et al. 2008). Single nucleotide polymorphisms (SNPs) in ARMS2 and HTRA1, two genes with strong linkage related to extracellular matrix function, are also associated with AMD susceptibility (Andreoli et al. 2009). Polymorphisms in the apolipoprotein E (apoE) gene modulate AMD risk as well (Thakkinstian et al. 2006). The E4 allelic variant seems to have a protective effect while the E2 allele is associated with increased AMD risk. Some recent SNPs in genes have suggested a role of cholesterol and lipid dysfunction in the development of the pathology. These include hepatic lipase gene (LIPC), cholesterylester transfer protein (CETP), lipoprotein lipase (LPL), and ATP-binding cassette, subfamily A member 1 (ABCA1).
Small RNAs have come into focus for their involvement in a number of developmental and disease states. Recent papers have suggested a role of microRNAs, small RNA processing enzyme, Dicer1, and Alu RNAs in the pathogenesis of AMD (Kaneko et al. 2011; Lukiw et al. 2012). NF-κB-regulated microRNAs are upregulated in both AMD and Alzheimer’s disease while other miRNAs including miRNA-9, miRNA-125b, miRNA-146a, and miRNA-155 are known to target 3′-UTR of CFH, thereby downregulating CFH. It has also been found that mir23a decreases in RPE cells from AMD donor eyes and is associated with cellular resistance to oxidative stress (Lin et al. 2011). Recent studies looking at small RNA processing have identified a reduction in the enzyme Dicer1, the small RNA processing protein, in RPE of patients with AMD (Kaneko et al. 2011). They however did not see a huge difference in microRNAs. Instead, they found a concomitant rise in Alu RNA accumulation in the cells. The group found that knocking-down DICER1 or overexpressing Alu RNA activates the NLRP3 inflammasome and triggers TLR-independent MyD88 signaling via IL18 in RPE cells (Tarallo et al. 2012). These could provide us with novel targets to treat the disease.
Glaucoma is one of the leading causes of vision loss and blindness, affecting as many as 60 million people around the world. It is estimated that nearly 80 million people will have glaucoma by the year 2020 (Quigley and Broman 2006). Like AMD, the pathogenesis of glaucoma is extremely complex and poorly understood. Glaucoma is characterized by progressing cupping of the optic nerve head, thinning of the nerve fiber layer, and retinal gliosis, all of which are associated with concomitant retinal ganglion cell death. The disease is classically associated with rise in the intraocular pressure. Dysfunction of the trabecular meshwork in the angle of the anterior chamber of the eye is linked with the disease development. There are two broad types: angle-closure glaucoma, where the angle of the eye is narrow preventing the outflow of the aqueous fluid, and open-angle glaucoma, in which case the meshwork itself is dysfunctional. In either case, there is rise in the intraocular pressure, which due to some yet unknown mechanism, leads to selective ganglion cell stress and loss. Additionally, there is a subset of patients with, what is referred to as, normal tension glaucoma. These patients have all the changes classically described for glaucoma in the absence of intraocular pressure changes.
A number of mechanisms have been postulated to explain the pathogenesis of glaucoma, including chronic intermittent ischemia due to rise in pressure, accumulation of reactive oxygen species, defective axon transport, loss of retrograde trophic factor support, and excitotoxicity (Calkins 2012). Defective axon transport is one of the earliest changes demonstrated in animal models of glaucoma. This could be due to mechanic pressure on the optic nerve head at the lamina cribrosa or be secondary to ischemia. This transport blockage also prevents retrograde transport of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and likely of other neurotrophic factors from central targets back to ganglion cells, which are critical for survival and dendritic maintenance. All together, these result in programmed cell death of the ganglion cells by upregulation of various proapoptotic markers. Several approaches have been utilized to identify genetic factors that are associated with glaucoma and multiple loci have been linked to open-angle glaucoma. Four genes with strong linkage include myocilin (MYOC), optineurin (OPTN), WD repeat domain 36 (WDR36), and neurotrophin 4 (NTF4) (Allingham et al. 2009). Genome-wide association studies have linked additional genes including caveolin 1 and 2 (CAV1/CAV2), transmembrane and coiled-coil domains 1 (TMCO1), and cyclin-dependent kinase inhibitor 2B (CDKN2B).
Diabetic retinopathy is one of the most common complications of diabetes and remains the leading cause of blindness among individuals in developed countries. Population-based studies suggest that approximately one-third of the population with diabetes have early signs of retinopathy and approximately one in ten has vision-threatening stages of retinopathy, such as macular edema and proliferative retinopathy (Chistiakov 2011). The major risk factors for diabetic retinopathy are hyperglycemia, high blood pressure, and duration of diabetes. Patients with diabetic retinopathy include the formation of microaneurysms, retinal hemorrhages, hard exudates, cotton wool spots, venous abnormalities, and neovascularization. The disease is associated with the loss of pericytes and endothelial cells, formation of acellular capillaries, thickening of the basement membrane, formation of microaneurysms, and neovascularization. The underlying mechanism on how hyperglycemia induces these changes is unclear but may involve changes in growth factor expression including VEGF and IGF1, activation of diacylglycerol–protein kinase C pathway, accumulation of advanced glycation end products, RAS activation, and misregulation of polyol pathway (Simo 2011).
Following degeneration in any of the aforementioned disorders, the retina needs to be rebuilt as innate repair mechanisms fall short of any visual improvement. Current avenues of research focus on two broad categories: (1) restoration of stem cell characteristics in host support cells to initiate some host repair and (2) replacement or generation of new cells in vitro which can then be transplanted to take over the function of lost cells.
15.3 Retinal Regeneration: Endogenous Mechanisms
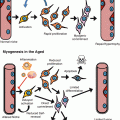
Amphibians, fish, birds, and mammals have all been shown to have varying degrees of retinal regenerative potential. This has been most prominent at the earliest stages of development and diminishes over time as the retina attains maturity (Del Rio-Tsonis and Tsonis 2003). Most retinal regeneration studies employ a process described as transdifferentiation, i.e., the ability of one cell type to take over a different fate. Transdifferentiation of neural retina involves three types of cells, support cells in the retina (called the Müller glial cells), cells from the RPE, and cells located in the ciliary epithelium/ciliary marginal zone at the periphery of the retina (Fig. 15.3). Regeneration via transdifferentiation involves three steps. First is the process of dedifferentiation where the RPE cells/Müller glial cells lose their native characteristics; second, they proliferate; and third and last, some of the cells differentiate into retinal cells (Del Rio-Tsonis and Tsonis 2003; Haynes and Del Rio-Tsonis 2004).
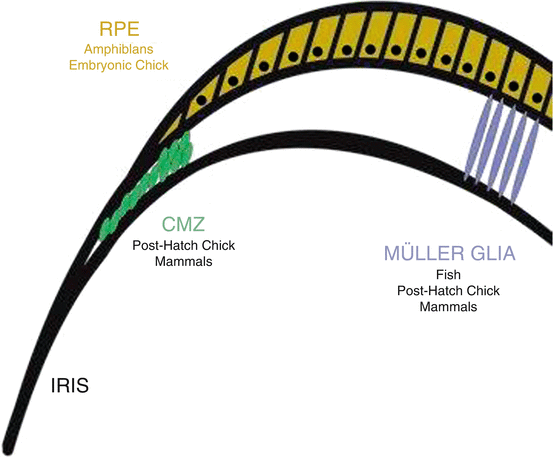
Fig. 15.3
Sources of endogenous retinal regeneration and repair. The figure highlights the three main sources of repair in the various species, i.e., RPE, CMZ, and Müller glia
15.3.1 Transdifferentiation from RPE Cells
Amphibians have a remarkable capacity to regenerate various organs and tissues including brain, limbs, and liver. Newt retina has been a great model to study neural retinal regeneration. Following removal of the entire neural retina, the eye can regenerate an entire complete and laminated new retina using the surrounding RPE cells (Stone 1950a, b; Stone and Steinitz 1957). The process involves proliferation and dedifferentiation of the RPE cells into an optic placode-like stem cell. These cells then regenerate a new bilayered structure, which has both a new neural retina and a new RPE cell layer. The newly formed retina has all the different cells including photoreceptors, bipolar cells, and ganglion cells as well as the inhibitory interneurons and glial cells. The only difference lies from the fact that the newly developed retina is inverted in orientation (Okada 1980). Recent studies have shown that the RPE transdifferentiation into neural cells occurs only in the presence of underlying connective tissue, i.e., choroid (Mitsuda et al. 2005). The results suggest the possibility of choroid playing an essential role in newt retinal regeneration potentially by means of diffusible substances. Two secretory factors that are upregulated in the choroid following newt retina removal include bFGF and IGF1. There is downregulation of Otx2, a key RPE gene, and upregulation of Notch pathway activity (Nakamura and Chiba 2007; Sakami et al. 2005). Some key regulators of this regeneration process include extracellular matrix components, such as laminin and heparin sulfate proteoglycans (Reh et al. 1987), as well as growth factors FGF-2 and IGF1 (Mitsuda et al. 2005; Sakaguchi et al. 1997), among others.
This phenomenon of RPE transdifferentiation also occurs in the embryonic chicken eyes (Coulombre and Coulombre 1965; Park and Hollenberg 1989; Pittack et al. 1991). When the neural retina is dissected out from the chicken embryos within the first 3–4 days of incubation (E3–E4), the RPE dedifferentiates and proliferates to form retinal progenitors, which then differentiate to form a new retina as in the newt. In the chick embryo, this process is stimulated by bFGFs with contribution from BMPs (Haynes et al. 2007) while activin signaling inhibits the regeneration (Sakami et al. 2008). Both FGFs and activin signaling are known to drive Pax6, a key transcription factor in retinal development. Recent experiments have shown that overexpression of Pax6 is sufficient to stimulate the production of neurons from the embryonic chicken RPE (Azuma et al. 2005). As opposed to the newt, in chickens, this RPE regenerative capacity is restricted to very early embryonic development. The mature chicken RPE does not contribute to retinal repair. So far, the data from mammalian RPE suggests very little contribution, if any, to retinal repair.
15.3.2 Transdifferentiation from Ciliary Margin
The peripheral margin of the retina where it meets the ciliary body has been thought of as another source of retinal stem cells and is called the ciliary marginal zone (CMZ). Retina of fish grows throughout life roughly matching the overall growth of the animal by the addition of new neurons from stem cells in the CMZ (Hollyfield 1968; Straznicky and Gaze 1971). In fact, most of the fish retina is generated from this CMZ as they grow to adult size. The CMZ cells resemble the embryonic retinal progenitor cells. The cells can generate clones of over 100 cells containing all the different retinal cell types (Wetts and Fraser 1988). Teleost fish also retain the ability to regenerate new retinal neurons following neurotoxin damage or surgical lesions throughout life (Hitchcock et al. 2004; Raymond et al. 2006). The chicken retina was largely believed to be completely developed at hatching. However, a study by Fischer and Reh showed that the CMZ is active in posthatch chickens (Fischer and Reh 2000, 2002). These cells at the retinal margin express genes common to embryonic retinal progenitors including Pax6. Injection of a combination of insulin IGF and EGF induces the cells in the CMZ to express progenitor markers and undergo proliferation. Similarly, sonic hedgehog induces proliferation in the adult chicken CMZ (Moshiri et al. 2005). A similar active CMZ has also been demonstrated in adult quails (Kubota et al. 2002).
Several reports have shown that cells isolated from the ciliary epithelium of several mammals, including mice, pig, and humans, can give rise to proliferative clones (neurospheres) when cultured in vitro (Ahmad et al. 2000; Asami et al. 2007; Coles et al. 2004; Kohno et al. 2006; MacNeil et al. 2007; Sun et al. 2006; Tropepe et al. 2000). The cells express a variety of neural progenitor markers and have been shown to undergo extensive proliferation. Pax6, neural progenitor gene, is required for the generation of these neurospheres (Xu et al. 2007). Upon exposure to differentiation conditions, these spheres were shown to express rod photoreceptors, bipolar and Müller glial genes. The sphere proliferation can be enhanced by exposure to Wnts, SCF, and PEDF (Inoue et al. 2006; Das et al. 2004, 2006a; De Marzo et al. 2010). Despite this early hope, some recent reports have suggested that these spheres may not completely resemble retinal stem cells. A detailed analysis by Mike Dyer’s group showed that all cells in the CMZ-derived spheres, including those expressing neural progenitor markers, were still pigmented and exhibited other characteristics of normal pigmented cells (Cicero et al. 2009). Thus, the jury is still out on the existence of a CMZ in humans, which can be exploited for native retinal repair.
15.3.3 Transdifferentiation from the Müller Glia
As discussed above, the fish retina contains a CMZ at the retinal margin, and it contributes to retinal growth as the fish increase in size. However, this zone is not thought to contribute to regeneration in central retina. Instead, fish contains two other quiescent progenitor populations, the rod progenitor cells and the Müller glia (Johns 1982). Upon injury, fish retinal Müller glia slowly start dividing to generate the rod progenitor cells (Bernardos et al. 2007). Using transgenic fish with GFP expressed in Müller glia, several labs have demonstrated that these cells are the primary source of regenerated neurons in fish central retina. Following damage to retinal neurons, the retina Müller glia reenter the cell cycle and dedifferentiate toward a progenitor-like phenotype (Raymond et al. 2006; Bernardos et al. 2007; Faillace et al. 2002; Fausett and Goldman 2006; Fimbel et al. 2007; Kassen et al. 2007; Thummel et al. 2008; Vihtelic et al. 2006; Wu et al. 2001; Yurco and Cameron 2005). A number of groups are now attempting to better understand the processes involved in reentry of the glial cells into cell cycle with the hopes of applying it to mammalian regeneration (Raymond et al. 2006; Kassen et al. 2007; Cameron et al. 2005; Yurco and Cameron 2007). It has been demonstrated that not all glial cells have the capacity to regenerate the retina but that these cells are asymmetrically distributed (Julian et al. 1998). Dan Goldman’s group has identified Ascl1, a basic helix-loop-helix gene expressed highly in retinal progenitors, as one that gets upregulated in Müller glia upon retinal damage (Fausett et al. 2008). They showed using morpholinos that inhibition of Ascl1 blocks Müller glial dedifferentiation and their subsequent reentry into the cell cycle. This is also interesting as a recent study in mammalian retina demonstrated the Ascl1 expression defines a subpopulation of lineage-restricted progenitors in the mammalian retina (Brzezinski et al. 2011). The process is mediated by EGF/MAPK/Notch cascade (Wan et al. 2012) and Wnt/β-catenin signaling (Ramachandran et al. 2011). A recent study looked for the inducer of Müller glial proliferation following damage. They found that TNF-α was produced by dying retinal neurons and was necessary for expression of ASCL1 in Müller glia (Nelson et al. 2013). Another secreted factor, which has been shown to induce glial proliferation in the absence of injury, is CNTF. It does this through a STAT3-dependent mechanism (Kassen et al. 2009). Injury-induced Ascl1 directly regulates lin-28, a microRNA-binding protein. Lin-28 then represses let-7 miRNA, which is normally involved in repression of regeneration-associated genes including ASCL1, PAX6, OCT4, KLF4, and c-MYC (Ramachandran et al. 2010). A detailed characterization study by Pam Raymond’s group into the genes involved in the zebrafish regenerative process showed that cells upregulated the Notch–Delta pathway as well as key retinal progenitor genes, Pax6a, Rx, and Chx10, upon reentry into the cell cycle (Raymond et al. 2006). These data suggest that regeneration and development use the similar molecular mechanisms and revoking a similar pathway in mammalian glial cells may restore regenerative capacity.
Like the fish retina, chicken retina displays some regenerative capacity (Fischer and Reh 2001). The earliest demonstration of this was using neurotoxic damage of inner retinal neurons with N-methyl-d-aspartate (NMDA) in posthatch chicks (Fischer and Reh 2001). Müller glia reenter the cell cycle and dedifferentiate into retinal progenitors (Fischer and Reh 2001, 2003; Fischer et al. 2002; Hayes et al. 2007). These Müller glial-derived retinal progenitors differentiate into amacrine cells, bipolar cells, and ganglion cells. There was no evidence of photoreceptor cell differentiation. A close examination of the Notch–Delta pathway revealed a dual role in the regenerative response (Hayes et al. 2007). Notch upregulation was indeed necessary for the glia to dedifferentiate and reenter cell cycle. However, after this initial need, Notch inhibition is necessary for differentiation of the Müller glial-derived retinal progenitors to form new neurons.
As opposed to fish and avian retinas, the mammalian Müller glia do not spontaneously reenter the cell cycle following retina damage (Chang et al. 2007; Close et al. 2006; Dayer et al. 2005; Sahel et al. 1990; Zhao et al. 2005). In order to get them to proliferate following damage, they require additional stimulation including growth factors. Ooto et al. used NMDA to induce damage in the rat inner retina (Ooto et al. 2004). They found that Müller glia underwent proliferation, though it was limited. They detected some of the Müller glial-derived progeny, which expressed markers of bipolar cells and photoreceptors. This number could be increased by co-injecting retinoic acid. Alternatively, overexpression of certain transcription factors including Math3, NeuroD1, Pax6, and Crx could generate bipolar and amacrine cells and photoreceptors. A number of additional publications have also reported similar BrdU-expressing photoreceptor cell generation following either retinal damage or growth factor treatment in adult rodents (Close et al. 2006; Das et al. 2006b; Osakada et al. 2007; Wan et al. 2007; Wan et al. 2008; Del Debbio et al. 2010). To gain a better handle on understanding the regenerative process in mouse retinas, Karl et al. conducted a detailed examination of the progeny derived from glia following toxic damage and EGF stimulation in various transgenic strains of mice to track various lineages (Karl et al. 2008). Using this protocol, up to 10 % of Müller glia entered the cell cycle within 3 days. They found that almost all of the Müller glial-derived progenitors differentiated to amacrine cells. Consistent with this observation, they found that FoxN4, a transcription factor necessary for the amacrine cell differentiation, was one of the most highly upregulated genes in both the mouse and chick retina after NMDA-induced damage. Other studies using Wnt3a or Shh following damage have reported photoreceptor cell differentiation (Osakada et al. 2007; Wan et al. 2007, 2008). There have been no studies of functional regeneration in either the chicken or the mammalian retina. Thus, it is still not known whether Müller glial-derived cells functionally integrate into the retinal circuitry.
Recently, the field of direct reprogramming has come into a lot of focus. Direct reprogramming or induced transdifferentiation refers to the process that allows cells to directly progress from one cell fate to another without an intermediate stem cell fate. The earliest landmark paper describing this was the direct conversion of fibroblast cells to myoblasts using a single transcription factor MyoD1 (Davis et al. 1987). More recently, a similar transdifferentiation strategy was used to generate neurons from fibroblast using ASCL1, BRN2, and MYT1L (Vierbuchen et al. 2010). A recent publication from Tom Reh’s lab attempted to employ the strategy to restore retinal stem cell characteristics to mammalian Müller glia (Pollak et al. 2013). As described above, Ascl1a is required for retinal regeneration in the fish and is rapidly upregulated in proliferating Müller glia. However, Ascl1 is not upregulated in the mouse retina after N-methyl-d-aspartate (NMDA)-induced damage (Karl et al. 2008). Therefore, the authors induced expression of ASCL1 in Müller glia in either dissociated cultures or explant retinas (Pollak et al. 2013). They found that ASCL1 was sufficient to induce neural differentiation of the glial cells. ASCL1 remodeled the chromatin at retinal progenitor gene sites activating their expression while at the same time downregulating glial genes. The reprogrammed cells expressed early postmitotic retinal markers including Otx2, Islet1, Neurod1, Prox1, and Crx. Thus, direct reprogramming could serve as an alternate strategy to induce retinal regeneration in mammalian retinas.
15.4 Retinal Repair: Cell Replacement
An alternative to finding ways to restore innate repair is to transplant fresh cells into the degenerated retina in order to restore visual function. Several potential sources for replacement neurons and photoreceptor cells have been hypothesized and tested. These include (1) dissociated differentiated retinal cells, (2) intact retinal sheets, (3) retinal progenitor cells, (4) neural progenitors derived from the CNS, (5) pluripotent stem cells, and (6) cells derived from CMZ and ciliary epithelium. The earliest published work on rodent retinal transplantation involved transplanting embryonic rat eyes into the brains and skin of newborn rats (Tansley 1946). Upon transplantation in skin pockets, only mesenchyme survived while the retina matured well in case of brain transplantations. Subsequently, a number of other intracranial transplantation studies were carried out by Ray Lund’s lab, to look at axon pathfinding. They showed that the axons of the transplanted cells can make contacts along the visual pathway and develop light responses (Lund and Hankin 1995; McLoon et al. 1981; McLoon and Lund 1980). The rats could also use functional visual cue from the graft site in various behavioral assays (Coffey et al. 1989; Klassen and Lund 1990). This early work showed that the mature nervous system is still plastic enough to accept new connectivity and information for functional tasks.
15.5 Transplantation of Retinal Cells
More recent work has concentrated on replacement of cells within the retina. There are two routes that could potentially be used to replace cells: (a) injection into the vitreal cavity close to the ganglion cell side and (b) injection into the subretinal space, potential space between the photoreceptors and overlying RPE cells. Integration following intravitreal transplantation has been limited due to inadequate migration of the transplanted cells into the retina probably due to tight junctions between astrocytes or end feet of Müller glia (Takahashi et al. 1998). Cell migration into the photoreceptor layer is more effective in normal adult retina if a subretinal approach is used (Lu et al. 2002; MacLaren et al. 2006). Developing retinal cells have been transplanted as intact retinal sheets (Seiler and Aramant 1998), retinal aggregates, or retinal spheres (del Cerro et al. 1991; Kwan et al. 1999) or as dissociated cell suspensions (MacLaren et al. 2006; Chacko et al. 2000; Qiu et al. 2005; Barber et al. 2013; Lakowski et al. 2011). Although retinal sheets and spheres survive and differentiate well in the subretinal space, the cells seem to self-aggregate into rosette and do not migrate much into the host retina from the graft site. However, enzymatically dissociating the developing retinas and transplanting cell suspensions results in better integration of the transplanted cells (Qiu et al. 2005).
The best demonstration of photoreceptor integration in a rodent retina was by MacLaren et al. (MacLaren et al. 2006); the authors dissociated retinal cells from a GFP-expressing transgenic mice wherein all the rod photoreceptors are green into the subretinal space of wild-type mice of different ages. They found that GFP-expressing cells migrating into the photoreceptor layer developed the morphology of mature photoreceptors. The cells upon integration elaborated outer segments and made synaptic connections with the host bipolar cells. The transplanted cells expressed markers of mature rod photoreceptors including rhodopsin. In the study, they compared integration from different donor ages varying from embryonic mice to adult mice. Interestingly, they found that the most successful integration was from newborn rod photoreceptors obtained from mice in the first postnatal week. Surprisingly, the most immature donor cells, from embryonic stages, were not effective for transplantation. These cells survived and matured but formed rosettes instead of integrating. A recent study from Tom Reh’s lab showed that adult mice can integrate just as well if their survival can be improved (Gust and Reh 2011). They found that the reason or poor integration was cell death due to the stress of dissociation and transplantation. Robin Ali’s group recently published a report comparing integration in various models of retinal degeneration ranging from slow to rapid degeneration (Barber et al. 2013). They found that a number of factors can influence integration efficiency including integrative of the outer limiting membrane of the retina formed by tight junction between glia, the gliotic status of the host retina, as well as speed of degeneration. Using approaches to alter the outer limiting membrane integrity just as ZO-1 siRNA or chondroitinase ABC can help enhance integration.
15.6 Transplantation of Non-retinal Cells
As discussed in the previous section, neurospheres can be generated from adult CMZ and ciliary epithelium of mammals (Ahmad et al. 2000; Asami et al. 2007; Coles et al. 2004; Kohno et al. 2006; MacNeil et al. 2007; Sun et al. 2006; Tropepe et al. 2000). In order to assess if these cells can be coaxed to differentiate into mature photoreceptor cells, Coles et al. transplanted ciliary-derived spheres into newborn mice by injecting them into the vitreal cavity of a newborn mouse and analyzing 4 weeks later (Coles et al. 2004). The authors saw migration of the transplanted cells to the retina and some integration into the photoreceptor layer as well as RPE. On account of the limited integration, the group recently tried to enhance integration and maturation by overexpressing two critical photoreceptor and RPE genes, OTX2 and CRX, while at the same time inhibiting CHX10, a progenitor gene (Inoue et al. 2010). They reported improved morphological integration with some functional improvement on behavioral and electrophysiological assays. A few groups have looked at neural progenitors as a source of replacement neurons (Takahashi et al. 1998; Young et al. 2000). These progenitors are obtained from the dentate gyrus of the hippocampus, a site of active neurogenesis even in adult mice. Hippocampal-derived neural progenitors exhibit a high degree of migration into all layers of the retina, following intravitreal injection in either newborn mice or adult mice with retinal degeneration or ischemia (Grozdanic et al. 2006; Guo et al. 2003; Akita et al. 2002; Kurimoto et al. 2001). Despite their hippocampal origin, they develop morphologies highly reminiscent of several types of retinal neurons; however, they did not show an ability to differentiate into mature photoreceptors.
15.7 Transplantation of Pluripotent Stem Cell-Derived Retinal Cells
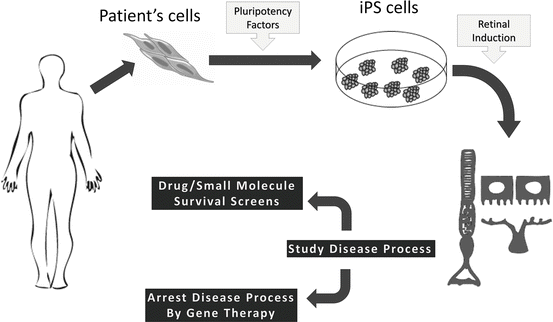
Pluripotent stem cells are most commonly known as cells with “blank slates,” having the potential to differentiate into any cell in the developed body. Currently under scientific investigation are two main groups of pluripotent cells: embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). ESCs are derived from the inner cell mass of a developing embryo at the blastocyst stage of development (Thomson et al. 1998), while iPSCs are pluripotent cells generated in vitro by the insertion and subsequent expression of a group of transcription factors: OCT4, KLF4, SOX2, c-MYC, LIN28, and NANOG (Fig. 15.4) (Takahashi et al. 2007; Takahashi and Yamanaka 2006; Yu et al. 2007). By virtue of pluripotency, these cells have the capacity to form the endoderm, mesoderm, and ectoderm germ layers seen in early development. Additionally, when grown under specific differentiation factors that help mimic the cellular environment of specific parts of the embryo, pluripotent cells can be urged to develop into specific cell fates that are seen in the developed body. Such a process that can be studied in vitro presents immense potential of investigating developmental biology, drug/toxicology studies, disease modeling, and aging, among other fields of study. Building on the knowledge of the network of eye-field transcription factors in the development of the vertebrate eye (Bharti et al. 2012; Zuber et al. 2003), scientists have been able to develop protocols with which retinal cells can be derived from pluripotent cells in vitro (Lamba et al. 2006, 2009a; Meyer et al. 2009; Osakada et al. 2008, 2009; Phillips et al. 2012; Hambright et al. 2012). This knowledge may play a further role in the development of practical and safe cell replacement treatments for retinal degenerative disorders.