Fig. 3.1
The loss of homeostatic apoptotic mechanisms promote neoplastic epithelial cell proliferation
3.3 Dose and Treatment Time
Skin cancer was the context for some of the earliest observations of clinically relevant radiobiology concepts. In 1936, Holthusen observed a sigmoidal-shaped distribution of the incidence of late normal tissue effects such as telangiectasia with increasing doses of radiation. A similar curve for skin cancer was derived and, thus, the idea of an optimal radiation dose for cure without complications [7], better known as the ‘therapeutic window’ (Fig. 3.2). In 1944, Strandqvist studied a single-centre series of patients irradiated for basal cell and squamous cell cancers of the skin and lip and became the first to establish a mathematical relationship between overall treatment time and response to ionising radiation [8].
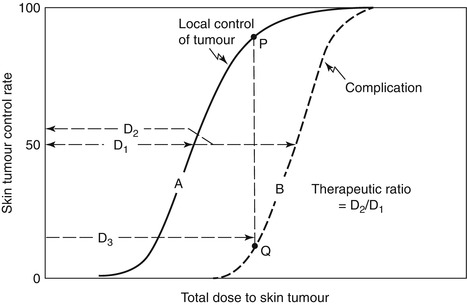
Fig. 3.2
The ‘therapeutic ratio’ or ‘window’ describes the separation between the probability of tumour control (solid line) and normal tissue complications (dashed line). In this theoretical example, the dose of irradiation required to achieve 50 % tumour control (D1) is less than that which will cause complications in 50 % of patients (D2). The addition of a radiosensitiser aims to reduce the dose of radiation necessary to achieve the same probability of tumour control and thus further reduce the probability of normal tissue damage and widen the therapeutic window (D1/D2)
3.4 The 5 Rs of Radiobiology
3.4.1 Repair
Multiple effective mechanisms have evolved to repair DNA damage and preserve genomic stability. After ionising radiation, DSBs may be repaired through homologous repair (HR), nonhomologous end joining (NHEJ) or, less commonly, by base excision repair (BER). 50 years ago, Strandqvist also observed that if radiotherapy was fractionated over several days, a greater total dose was required to be equivalent to a lower dose given in a single treatment. Part of this is due to the ability of tumour cells to repair damage between fractions, which reduces the efficacy of treatment. DNA repair is a double-edged sword however, as it also enhances normal tissue tolerance.
3.4.2 Repopulation and Fractionation
Tumour cell death is the desired outcome of radiotherapy; however, a margin of 5–10 mm of normal skin is added to the visible extent of, for example, a basal cell or squamous cell skin cancer, both laterally and at depth, to cover the spread of any microscopic tumour cells that may cause a tumour recurrence. This margin should receive 90–95 % of the prescribed dose, and adjacent normal epithelial cells will therefore receive a cytotoxic and genotoxic dose of ionising radiation. Cell proliferation is a normal response to epithelial damage, and stem cells divide and differentiate to replenish the epithelium. Dry desquamation is a sign of increased cell loss, but when moist desquamation occurs, the rate of repopulation of the epithelium from stem cells in the basal layer is being exceeded [9, 10]. Tumour cell repopulation will also occur during a course of fractionated radiotherapy. Even more unfortunate is that radiation can activate cell signalling networks that promote accelerated repopulation. This is especially true for squamous carcinomas. Tumour cell repopulation is an important mechanism of treatment failure and inhibitory strategies are evolving [11].
As epithelial tissues proliferate rapidly, there is no latency in the development of acute effects. Human skin has a high repair capacity and early skin radiation reactions display a half-time to recovery of only 1 hour [12]. Skin thus has a high fractionation sensitivity and is attributed a high α/β ratio, a radiobiology concept to describe a tissue’s response to radiation based on the linear quadratic equation [13]. Acute reacting tissues, such as skin and gastrointestinal epithelia, have a high α/β ratio (approximately 10) [14]. In contrast, late responding tissues such as the spinal cord, where DNA damage is only displayed at infrequent cell divisions, have a low α/β ratio (approximately 2). Radiosensitive non-melanoma skin cancer has an α/β ratio of 6–10, whilst melanoma cells typically have a lower α/β ratio, in the order of 3. These α/β ratios can be exploited when choosing the optimal fractionation schedule.
3.4.3 Reassortment
Normal cells divide by progressing through the cell cycle. Quiescent cells (G0) may be recruited into the cell cycle in response to proliferative signals. The cyclin/cyclin-dependent kinases (CDKs) form complexes that act at each phase of the cell cycle to promote entry into the subsequent phase, but natural CDK inhibitors regulate this. The G1 and G2 checkpoints provide an opportunity for the normal cell to audit the integrity of the DNA before further synthesis and cell division, respectively. The tumour suppressor gene p53 is requisite for G1 arrest [15]. As loss of p53 is the most common genetic aberration in cancer, tumours frequently have deregulated cell cycle control and are able to bypass these checkpoints and proliferate unchecked.
Reassortment is the normal redistribution of cells through the cell cycle. As cells move through the cell cycle, it is plausible that cells that were in the relatively radioresistant S phase of the cell cycle during a fraction of radiotherapy will have moved into G2 and M phases and be more sensitive to radiation for subsequent fractions [16].
3.4.4 Reoxygenation
A tumour consists not only of tumour cells but also a stromal matrix that includes, perhaps most significantly, endothelial cells. The ability of a tumour to establish its own vasculature is prerequisite for its survival beyond a cluster of cells greater than 100 μm in diameter [17, 18]. To achieve this, tumours increase secretion of pro-angiogenic proteins such as vascular growth factors (VEGFs) relative to the production of anti-angiogenic factors, including angiostatin, endostatin and thrombospondin-1.
Endothelial cells express high levels of the family of vascular endothelial growth factor receptors (VEGFRs). In endothelial cells in vitro, ionising radiation has been shown to activate VEGFR directly, and consequently AKT, resulting in a possible pro-angiogenic and pro-survival effect. VEGF-mediated radioresistance [19], through the upregulation of the anti-apoptotic protein bcl-2, is a significant clinical problem but one that can be inhibited with targeted agents.
Rapidly proliferating squamous cell carcinomas of the skin are liable to outgrow their blood supply and become necrotic and hypoxic in the centre. Following a fraction of radiotherapy, the oxygenated cells will be killed and there will be a higher percentage of hypoxic cells. Following mechanisms that include the opening of temporarily closed vessels, reduced respiration in damaged cells and mitotic death, the hypoxic fraction will return to pre-radiation levels which is termed re-oxygenation [20].
3.4.5 Intrinsic Radiosensitivity
Cutaneous malignancies display a particularly wide spectrum of intrinsic sensitivity. The main reason appears to be the extent and facility of tumour cell apoptosis, as deregulation of this cell death pathway renders tumour cells resistant to conventional therapy. It has been demonstrated that tumours grown in p53-deficient mice display a lower proportion of apoptotic cells and fail to regress in response to chemotherapy or gamma irradiation, as compared with those with functional p53 [21] (Fig. 3.3). p53 is one of several compelling targets in cancer therapy, especially in relatively resistant tumours such as melanoma.
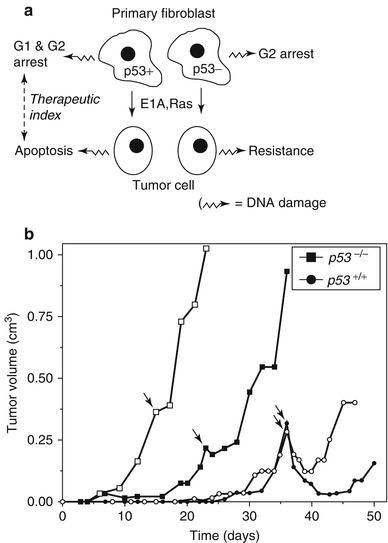
Fig. 3.3
p53 is a key mediator of intrinsic radiosensitivity. (a) In this in vitro example using primary fibroblasts, p53-deficient cells subsequently underwent oncogenic transformation, for example, by Ras mutation. In contrast to the p53 wild-type cells, the p53-deficient cells become resistant to DNA damage by irradiation. (b) In vivo, p53 wild-type xenografts from the oncogenically transformed fibroblasts displayed enhanced radiosensitivity following irradiation with 7 Gy relative to p53 mutants. The white and black shading represents different clones
Patients diagnosed with the recessive condition ataxia telangiectasia have mutated ATM/ATR genes and may show exquisite sensitivity to radiotherapy. Where ataxia telangiectasia was unsuspected, excessive normal tissue toxicity has resulted from standard radiotherapy schedules. A reduction in both DNA repair fidelity and G1 delay may underlie this sensitivity, and impaired DSB repair has also been implicated. Patients with breast cancer found to have ATM gene mutations did not display increased radiation-induced skin toxicity, however [22]. In theory, assessing the intrinsic sensitivity of a patient’s tumour or fibroblasts may enable personalisation of the radiotherapy dose prescription. However, a study in breast cancer has suggested that beam energy and treated volume may impact more on late effects than the intrinsic radiosensitivity of fibroblasts [23].
3.5 Clinical Applications
Lymphoma and melanoma are classic examples of the potential extremes of the spectrum of tumour intrinsic radiosensitivity. Basal cell and squamous cell carcinomas lie towards the more sensitive end of the range. The marked difference in clinical response to radiotherapy has caused contrasting radiotherapy schedules to evolve for the three classes of skin tumour, to best exploit the underlying differences in radiobiology.
3.5.1 Radiosensitive Tumours
3.5.1.1 Lymphoma
The tendency of lymphoma cells to undergo apoptosis readily means that radiotherapy is a very effective treatment for cutaneous T- and B-cell lymphomas. It is unnecessary to exploit particular radiobiological features with the exception of intrinsic sensitivity, which allows a marked dose reduction. Even very low-dose radiotherapy (4 Gy in two 2 Gy fractions) can achieve complete remission of a low-grade small cutaneous lesion [24]. Across the various types of lymphoma, prognosis is generally good and a lower dose of radiation will be associated with a lower risk of radiation-induced second malignancy. The consequent normal tissue sparing is also important in a disease where chemotherapy may play an important role and toxicities of treatment may overlap.
3.5.1.2 Basal and Squamous Cell Carcinoma
As discussed, squamous cell skin cancers in particular can repair DNA and repopulate during a 6-week course of radiotherapy. Reducing overall treatment time (by increasing fraction size, treating 6 days a week or compensating for any missed fractions) is therefore very useful to counteract accelerated repopulation [25]. As an early reacting tissue, the late effects of radiation on the skin (fibrosis, atrophy and telangiectasia) vary less as a function of fraction size than late reacting tissues. For a squamous cell carcinoma of less than 5 cm diameter, a typical curative radiation prescription of 66 Gy in 33 fractions at 2 Gy per fraction over 6 1/2 weeks may by accelerated to 55 Gy in 20 fractions over 4 weeks or 45 Gy in 10 fractions over 2 weeks with equivalent efficacy and no significant increase in long-term complications [26].
Normal cells repopulate an area of irradiated skin from the periphery, thus the diameter of the radiation field and consequently the 3-D volume are important to consider when planning radiotherapy and counselling the patient about anticipated side effects. The prescribed dose is also a significant factor as the higher the dose, the slower the normal tissue recovery due to the greater stem cell depletion.
3.5.2 Radioresistant Tumours
Within skin tumours, malignant melanoma is generally considered to display the greatest relative intrinsic radioresistance. Consequently, surgery is established as the current most effective clinical treatment modality for primary tumours. The range of radiation sensitivity observed in human tumour xenografts following single-dose irradiation may be very variable, however [27], reinforcing radiotherapy as a useful treatment modality. In metastatic melanoma, palliative hypofractionated radiotherapy may be offered using a large dose per fraction (20 Gy in 5 fractions over 1 week or 36 Gy in 6 fractions weekly for 6 weeks). Given the frequently poor response of melanoma even to hypofractionated radiotherapy, efforts have been made to exploit the tumour radiobiology to improve patient outcome through the administration of radiosensitisers. The ideal radiosensitiser increases tumour cell kill without adding to normal tissue toxicity at the same dose of radiation, thus it widens the therapeutic window.
3.5.3 Strategies to Overcome Intrinsic and Acquired Radioresistance
Whilst melanoma may demonstrate intrinsic radioresistance, recurrent previously treated squamous cell carcinoma of the skin is often more resistant to therapy than at first presentation. This is usually due to the survival of the most resistant cells, which then proliferate to cause a recurrence. Such refractory cells often have clonogenic, stem cell-like properties which may also contribute to accelerated repopulation [28], and current research efforts are directed at identifying and eradicating these. With time, surviving tumour cells may acquire further genetic mutations that increase radioresistance.
3.5.3.1 Chemotherapy
The addition of cytotoxic chemotherapy agents to radiation can induce apotosis if the tumour remains ‘prone’ with acceptable normal tissue toxicity. This is standard practice in other moderately sensitive epithelial cancers such as cervix, anal/colorectal and squamous cell head and neck cancers, for example. Standard chemotherapy has not proven to be an effective radiosensitiser in melanoma, however.
3.5.3.2 Molecular Targeted Therapy
Molecular targeted agents are a more novel class of radiosensitiser. The most successful example is a monoclonal antibody to the epidermal growth factor receptor (EGFR). Cetuximab is now frequently prescribed as a radiosensitiser where patients with squamous cell carcinoma of the head and neck (SCCHN) are unsuitable for the standard chemotherapy drug. Radiation can activate EGFR directly, as well as increasing the release of cognate ligands and reducing inhibitory phosphatase activity, as a response to the cellular stress. EGFR signalling drives cells through the cell cycle and reduces apoptosis, thus contributing to accelerated repopulation. The role of EGFR inhibition in the therapy of melanoma is under evaluation [29].
3.5.3.3 Promotion of Apoptosis
Due to the low intrinsic propensity for apoptosis in melanoma, the combination of p53 inhibitors with radiation is, perhaps, a more compelling strategy. Ionising radiation may trigger signalling through p53 to Mdm2 which precipitates the degradation of p53 and reduces apoptosis. The nutlins are a family of molecules that can prevent the interaction of p53 with Mdm2 and thus preserve p53 function, and these are starting to be combined with radiation [30]. E2F1 is a transcription factor that can induce apoptosis independent of functional p53, which is an interesting target as p53 is often deleted or mutated in cancer [31]. Recently, knockdown of E2F1 gene expression was demonstrated to reduce EGFR expression and reduced the invasive, but not proliferative, capacity of melanoma cells [32]. However, the Mdm2 and E2F1 pathways form part of a signalling network and thus inhibition of both in combination with radiotherapy has proven more effective in experiments on prostate cancer cell lines [33]. This rationale should hold true for melanoma.
Furthermore, there is clinical evidence of the feasibility of using recombinant proapoptotic ligands to drive receptors capable of activating p53-independent apoptosis, such as rhApo2L/TRAIL. Monoclonal antibodies to the death receptors DR4 and DR5 on the extrinsic pathway have reached phase II clinical trials, and TRAIL receptor antibodies are being tested in combination with radiotherapy [34




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
