Fig. 22.1
Cellular components of healthy and inflamed skin. Representative immunohistochemistry of normal appearing, nonlesional skin of psoriasis patients, and lesional psoriasis skin, and summary of immune cellular components and their surface receptors. With hematoxylin and eosin stain (H&E), lesional psoriasis skin shows a greatly thickened epidermis (acanthosis) with elongations into the dermis (rete ridges). Retention of nuclei (parakeratosis) can be seen in the thickened stratum corneum. There is a dramatic increase in the number of cells in the dermis, composed predominantly of dendritic cells (DCs) and T cells. There are increased CD3+ T cells in lesional psoriasis skin, often forming lymphoid-like clusters with DCs. Keratin 16 (K16) stains basal epidermis in nonlesional skin, but full thickness epidermis in psoriasis. In the steady state, CD11c+CD1c/ blood dendritic cell antigen (BDCA)-1+ resident myeloid DCs (mDCs) are found in the upper dermis in nonlesional skin. During psoriatic inflammation, there is an increase in CD11c+ inflammatory mDCs in the epidermis and the dermis that are CD1c/BDCA-1–. These cells express HLA-DR, TRAIL, and Toll-like receptor (TLR)1/2. Among increasing CD11c+ DC populations, tumor necrosis factor (TNF) and inducible nitric oxide synthase (iNOs)-producing DCs (TIP-DCs), which express high levels of TNF and iNOS, are also found. In addition, CD208/DC-lysosomal–associated membrane protein (DC-LAMP)+ mature DCs exist in the dermis, forming aggregates with CD3+ T cells. Resident mDCs are stable in number between nonlesional and lesional skin, and are both CD11c+ and BDCA-1+. CD207/langerin+ Langerhans cells are found scattered in the lower epidermis in nonlesional skin and are found higher up in the thicker epidermis in lesional skin. Langerhans cells are also identified by CD1a and HLA-DR. All images 10× magnification
In the dermis, there are abundant mononuclear cells, predominantly CD3+ T cells and CD11c+ dendritic cells (DCs). Psoriasis lesions contain prominent aggregates of intermixed T cells and CD208/DC-lysosomal–associated membrane protein (DC-LAMP) + mature DCs in the dermis. Langerhans cells exist mainly in the upper part of the epidermis, showing redistribution. In lesional skin, a greater number of dilated dermal blood vessels are seen. Endothelial cells are activated in psoriatic lesions, as is indicated by staining for intracellular adhesion molecule-1(ICAM-1, also known as CD54), vascular cell adhesion molecule-1 (VCAM-1, or CD106), and E-selectin (CD62E). Leukocytes can gain entry to skin parenchyma by transmigration through reactive vessels, but resident skin leukocytes might also expand to create the dense infiltrates seen in psoriatic lesions.
For many years, there was a debate whether the primary process in psoriasis involved hyperplastic keratinocytes with secondary immune activation or vice versa. In part, this debate was fueled by lack of knowledge regarding therapeutic mechanisms of commonly used agents. Corticosteroids and some immunosuppressants could be used to treat psoriasis, but on the other hand, systemic agents such as methotrexate were viewed as keratinocyte-directed agents. Although cyclosporine, a calcineurin antagonist, had a dramatic effect for disease activity, this agent has direct effects on keratinocytes and immune cells, so an immune pathogenesis could not be proven solely with this agent [12, 25]. The first specific indication that the immune system could be playing a more integral role came with the clinical trial targeting T cells with the DAB389IL-2 agent , a fusion protein also called denileukin diftitox, that causes apoptosis in activated T cells expressing functional IL-2 receptors [23]. This study showed that specific depletion of activated T cells in psoriasis lesions could cause clinical and histological disease resolution.
Hence, the DAB389IL-2 study set up the general hypothesis that psoriasis is a disease mediated by activated T cells that are present in focal skin regions. This view has been solidified and refined by the availability of a series of immune-targeted drugs that have been tested in psoriasis patients (Table 22.1). CTLA4-Ig (abatacept) was used to block B7-mediated costimulation to T cells [1]. At high doses, consistent improvements in psoriasis were detected that correlated with depletion of DC and T cell subsets from diseased skin regions. Therefore, this study was the first to show that disease activity could potentially be restrained by a specific T-cell antagonist that did not deplete T cells as its primary mechanism of action. Subsequently, two biologics targeted to T-cell activation pathways became FDA approved therapeutics for psoriasis. One of these agents was an LFA-3-Ig fusion protein (alefacept) that blocks CD2-mediated T-cell activation. With this agent, strong clearing of psoriasis lesions was seen in a subset of patients where the drug induced large decreases in T cells and DC populations in the skin [7]. Memory T cells were often depleted in the peripheral circulation of patients treated with alefacept [6], providing additional evidence for pathogenic actions of activated T cells that infiltrate skin lesions of psoriasis.
Table 22.1
Systemic therapeutics for psoriasis
Target | Agent | Drug |
|---|---|---|
General T-cell immunosuppressive agents tested or used in psoriasis treatments with known molecular target | ||
Calcineurin | Cyclosporin | Neoral |
IL-2R | DAB389IL-2 | Denileukin diftitox |
CTLA4 | CTLA4-Ig | Abatacept |
LFA-3 | LFA-3-Ig | Alefacept |
CD11a | Anti-CD11a | Efalizumab |
Protein kinase C (PKC)θ | PKC inhibitor | AEB071 |
(Sotrastaurin) | ||
Phosphodiesterase type 4 (PDE4) | PDE4 inhibitor | CC-10004 |
(Apremilast) | ||
Cytokine targeted agents or therapies for psoriasis | ||
TNF | Anti-TNF | Infliximab |
Adalimumab | ||
TNF | TNFR-Ig | Etanercept |
IL-12/23 | Anti-IL-12/23p40 | Ustekinumab |
Briakinumab | ||
IL-23 | Anti-IL-23p19 | LY2525623 |
SCH 900222 | ||
CNTO 1959 | ||
AMG 139 | ||
IL-17A | Anti-IL-17A | Secukinumab |
Ixekizumab | ||
IL-17RA | Anti-IL-17RA | Broadalumab |
Another T-cell targeted biologic approved for use in psoriasis was a monoclonal antibody to the integrin CD11a (anti-CD11a , efalizumab). T cells selectively use the integrin CD11a/CD18 (LFA-1) for migration to peripheral tissues and as a part of T-cell costimulation. Efalizumab blocks T-cell migration and activation responses in psoriasis patients, again without inducing T-cell cytotoxicity as a primary mechanism. Strong improvements in psoriasis lesions were seen in patients that had accompanying reductions in T-cell and DC subsets that infiltrate skin lesions [38]. Subsequently, the role of different T-cell subsets in psoriasis, including Th1, Th17, and Th22, has been dissected through testing of a range of cytokine antagonists [47], listed in Table 22.1.
22.4 Effector Immune Pathways in Psoriasis
In the early 2000s, the Th1 pathway was regarded as the dominant pathogenic model for psoriasis [35]. At that time, it was appreciated that there was a strong interferon (IFN)-γ signature in psoriasis lesions: IFN-γ–producing Th1 cells were abundant in psoriasis lesions and blood, and these Th1 cells were reduced with successful therapy. Recently, clinical studies have been conducted with individual cytokine antagonists that suggest a central role for IL-23 and IL-17, along with TNFα, in driving disease pathology. Therefore, the current pathogenic model of psoriasis emphasizes the IL-23/T17 axis, but also contains other T-cell subsets. For this chapter, IL-17–producing T cells are denoted as T17.
In the steady state, there are three DC populations residing in the skin: Langerhans cells, resident dermal myeloid DCs (mDCs), and plasmacytoid DCs (pDCs) . During psoriatic inflammation, both mDCs and pDCs increase in number, but the greater increase comes from the mDC population [46, 71]. Blood dendritic cell antigen (BDCA)-2+CD123+ pDCs have been proposed to have an important role in the triggering of lesions, although mDCs are appreciated to be key proximal cells in the pathogenic psoriatic pathway. Psoriasis can be triggered by many factors, including injury and trauma (termed the Koebner effect), infection, medications, and the topical biological response modifier Imiquimod (Fig. 22.2a). Injury to the skin may cause cell death and increase production of the antimicrobial peptide LL37 by keratinocytes. DNA/LL37 complexes bind to intracellular Toll-like receptor (TLR)9 in pDCs, which causes activation and production of type I interferons, IFNα/β. LL37/RNA complexes can activate pDCs through TLR7, and mDCs can be activated by the LL37/RNA complex as well as by type I interferons, driving T-cell activation and the production of cytokines found in psoriasis [17, 19, 31]. In addition, murine studies have shown that topical Imiquimod (a TLR7 agonist) may induce psoriasiform skin inflammation, mediated by the IL-23/IL-17 axis and activated DCs [64]. mDCs existing in the skin are generally characterized using the classic DC definition, CD11c+HLA-DR+ cells. The major resident population of mDCs in normal dermis is identified phenotypically with a single monoclonal antibody, CD1c, which is also known as BDCA-1. In the steady state, CD1c/BDCA-1+ mDCs are relatively immature with modest T-cell stimulatory ability [70]. However, many CD11c+ DCs in psoriasis lesions express markers of DC maturity, such as CD208/DC-LAMP and CD83 [38], thus they could function as conventional DCs in terms of presenting antigens to T cells for triggering of acquired immune responses. During psoriasis inflammation, there appears to be an additional mDC population that did not costain BDCA-1, and were subsequently termed inflammatory mDCs or TNF- and inducible nitric oxide synthase (iNOS)-producing DCs (TIP-DCs) [68]. They are likely derived from circulating precursors migrating into the skin due to inflammatory and chemotactic signals. Inflammatory mDCs express inflammatory molecules that include TRAIL, TLR1, TLR2, and S100A12 [69]. TIP-DCs express high levels of TNF and iNOS, thus referred to as TIP-DCs [38]. Pathogenicity of TIP-DCs in psoriasis is suggested by the rapid downmodulation of TIP-DC products TNF, iNOS, IL-20, and IL-23 during treatment with effective therapies [26, 67]. TIP-DCs can also stimulate the differentiation and activation of Th17 T cells [68]. Activated DCs in psoriasis produce IL-23 and IL-12 [68], which stimulate the three populations of resident T cells, T17, Th22, and Th1 cells. IL-23 activates T17 cells to produce IL-17A and IL-17F, which drive keratinocyte responses. mDCs also produce IL-20 in psoriasis lesions [65], and this could be a driver of epidermal hyperplasia. Interestingly, both resident (BDCA-1+) and inflammatory (BDCA-1–) mDCs have been shown to induce the proliferation of T cells and induce allogeneic T cells to produce IFN-γ and IL-17 [68], which are abundant in lesions [39].
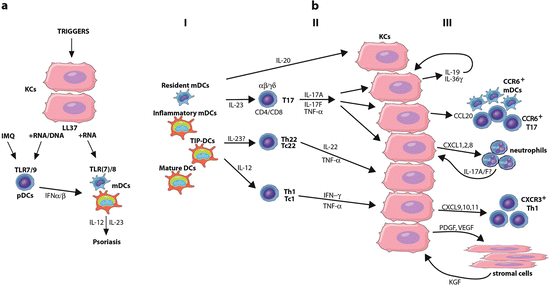
Fig. 22.2
Pathways for initiation and maintenance of psoriasis. (a) Initiation phase. Imiquimod (IMQ), a Toll-like receptor (TLR)7 agonist, can activate plasmacytoid dendritic cells (pDCs) to produce interferons (IFNs). LL37, a peptide derived from cathelicidin, may have an important role in the initiation of psoriasis lesions via the pathway. LL37 released from keratinocytes (KCs) can bind to nucleic acids to activate pDCs to release IFNα/β. LL37/RNA complexes can also activate myeloid DCs (mDCs) to produce IL-12 and IL-23, key psoriatic cytokines. (b) Maintenance phase (chronic disease). Major pathogenic pathway in psoriasis with (I) resident and infiltrating DCs producing cytokines such as interleukin (IL)-23 and IL-12. (II) These cytokines activate IL-17–producing T (T17), T helper (Th)22, and Th1 cells, to contribute to the cytokine milieu and further act on KCs. (III) Upon activation, KCs can produce chemokines to attract DCs, T cells, and neutrophils to the skin. Cytokines produced by KCs act as keratinocyte autocrine and/or paracrine growth factors. TNF tumor necrosis factor, TIP-DCs TNF and inducible nitric oxide synthase-producing dendritic cells, PDGF platelet-derived growth factor, VEGF vascular endothelial growth factor, KGF keratinocyte growth factor
Whereas keratinocytes might be viewed only as bystander cells in terms of immune activation, it is more likely that they are active participants in the recruitment and activation of leukocytes in psoriasis lesions (Fig. 22.2). IFN-γ induces keratinocytes to produce an array of pro-inflammatory chemokines, including CXCL9, 10, and 11, to recruit more CXCR3+Th1 cells into the lesion [48]. By contrast, IL-17 induces the expression of chemokines (CXCL1, 2, and 8) that are involved in neutrophil recruitment into the site of inflammation. Aside from neutrophil-attraction chemokines, IL-17 induces the elaboration of CCL20, which recruits more CCR6+CD11c+ mDCs and CCR6+ T17 T cells from peripheral circulation thus potentially creating a self-amplifying loop of inflammation [48]. LL37, which is also induced by IL-17 [54], may establish another self-amplifying inflammatory loop by forming complexes with self-RNA, and inducing mDCs to mature and express DC-LAMP [17]. Furthermore, activated T17 cells also elaborate TNF-α [39], which has synergistic effects with IL-17 as they share common transcriptional regulatory elements (NF-κB and CCAAT/enhancer-binding protein) [57]. IL-17 induces IL-19 and IL-36γ in psoriasis lesions, which may then lead to proliferative responses in keratinocytes. IL-19, IL-20, and IL-22 have similar trophic effects on the epidermis [55], and transgenic models have shown psoriasis-related pathologies in mouse skin for IL-20, IL-22, and IL-36 cytokines, reviewed in [40]. In humans, Th22 T cells are the main producers of IL-22. IL-17 and IL-22 cooperatively enhance the expression of epithelial antimicrobial peptides [36]. In addition, keratinocyte-derived cytokines such as platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) influence the growth of supporting stromal cells. Activated stromal cells overproduce factors such as keratinocyte growth factor (KGF) that can induce proliferation of keratinocytes [14].
After DC activation, activation and differentiation of T-cell subsets are supported by IL-12 and IL-23 , which are produced mainly from mDCs in the skin. Psoriasis lesions contain T cells that discretely produce IFN-γ, IL-17, and IL-22, with initial labeling of these cells as Th1, Th17, and Th22, respectively. There are also CD8+ T cell populations that make the same range of cytokines, thus these have been termed Tc1, Tc17, and Tc22, respectively. More recently, γδT cells have been found to be IL-17–producing cells in psoriasis, so we have adopted the more general term T17 to encompass IL-17–producing lymphocyte subsets in the skin. Figure 22.2b shows a current pathogenic model for the involvement of T cells and DC subsets in sustaining disease activity in psoriasis.
22.5 Therapeutic Agents Target Activated Immunity in Psoriasis
Current therapeutic options for psoriasis include various topical agents for limited disease (corticosteroids and vitamin D analogues), oral systemic immunosuppressive agents (such as methotrexate, retinoids, and cyclosporine), and phototherapy. More recently, biologics such as anti-TNF and anti-IL-12/23 antibodies have been FDA approved, and there are evolving agents such as anti-IL-23p19 and anti-IL-17 currently in clinical trials (Table 22.1). Therapeutic effectiveness of various agents may be related to how well this cycle of inflammation is broken.
22.5.1 New T-Cell Targeted Agents
Anti-inflammatory agents, including protein kinase C (PKC) inhibitor and phosphodiesterase (PDE)4 inhibitor are also in clinical trials. PKC isoforms have been shown to play key roles in cellular signaling, proliferation, differentiation, migration, survival, and death. PKCα and PKCθ as well as PKCβ and PKCδ are functionally important for T and B cell signaling, respectively [24, 42, 59]. PKCθ plays an essential role in T-cell activation because it is the only isoform that is selectively translocated to the T-cell/antigen-presenting cell contract site immediately after cell–cell interaction [43]. Furthermore, PKCθ is crucial for IL-2 production, a prerequisite for the proliferation of T cells [2]. AEB071 (sotrastaurin) is a novel PKC inhibitor that has strong and specific activity on PKCθ, PKCα, and PKCβ and lesser activity on PKCδ, PKCε, and PKCη. Actually, AEB071 both abolishes the production of several cytokines by activated human T cells, keratinocytes, and macrophages in vitro and inhibits an acute allergic contact dermatitis response in rats [58]. Clinical severity of psoriasis patients who were administered AEB071 was reduced up to 69 % compared with baseline after 2 weeks of treatment [58]. The improvement in psoriasis patients was accompanied by the histological improvement of skin lesions and may be partially explained by a substantial reduction of p40+ dermal cells [58], which are known to mediate psoriasis.
PDEs comprise a family of enzymes that uniquely hydrolyze and degrade cyclic adenosine monophosphate (cAMP). One of 11 subtypes, PDE4 is widely expressed in numerous cell types, including hematopoietic cells, keratinocytes, endothelial cells, and nerve cells [9, 27]. Among its various cellular functions, PDE4 regulates immune and inflammatory processes through control of intracellular cAMP levels and downstream protein kinase A pathways [50]. With PDE4 inhibition, and the resulting increases in cAMP levels in immune and nonimmune cell types, expression of a network of pro-inflammatory and anti-inflammatory mediators can be modulated. In T cells, elevated levels of cAMP decrease the intracellular signaling triggered by the CD3-TCR complex on the cell surface. CC-10004 (apremilast) is an orally available PDE4 inhibitor that modulates a wide array of inflammatory mediators involved in psoriasis, including decreases in the expression of iNOS, TNF-α, IFN-γ, IL-12, IL-23, CXCL9, and CXCL10 and the increase in IL-10 [3, 5, 56]. A Phase II open-label study in recalcitrant plaque psoriasis showed that mean percent decreases (improvements) from baseline in psoriasis patients administered apremilast were −59 % for PASI score and −53 % for body surface area at week 12. Skin samples taken from patients after treatment showed significant reductions of CD11c, CD3, and CD56 positive cells [22]. It indicates that CC-10004 reduced mDC, T-cell, and NK-cell or NK-T cell infiltration into the epidermis and the dermis. Reduced inflammatory leukocytes, with a pattern of broad partial inhibition, suggested reduced IL-23/T17 and Th22 response pathways.
22.5.2 Specific Cytokine Antagonists as Therapeutics
TNFα blockade was the first widely used anticytokine therapy for psoriasis. In psoriasis, TNFα is produced by keratinocytes, DCs (particularly TIP-DCs), Th1 cells, Th17 cells, and Th22 cells [13, 38, 39]. Blockade of TNFα is very efficacious, with >50 % of subjects achieving a psoriasis area and severity index (PASI)75 by 3 months. There are three FDA-approved anti-TNFα agents approved to treat psoriasis: infliximab is a chimeric monoclonal antibody that binds soluble and membrane TNF; etanercept is a soluble TNFα receptor-IgG fusion protein; and adalimumab is a fully human anti-TNFα IgG1 monoclonal antibody. Insights to the mechanism for the effective therapy with TNF modulators come from genomic studies that follow psoriasis lesions with etanercept treatment. These studies found that disease improvement correlated with the rapid downmodulation of IL-23 and Th17 cell products, and final disease resolution correlated with the late downmodulation of Th1-associated genes [67, 72]. Furthermore, comparison of responders and nonresponders to etanercept treatment revealed that successful response to treatment was dependent on the inactivation of mDC products and subsequently the Th17 pathway [72]. Thus, the TNFα blockade may be linked to suppression of the IL-23/Th17 axis.
To date, direct blockade of IL-17 appears to be the antipsoriatic therapeutic strategy with the most rapid efficacy. The studies presented below suggested that IL-17 ligands are the key pathogenic psoriatic cytokines, and that inhibiting this axis is essential for disease resolution. IL-17 and its downstream genes are turned off by many other psoriasis treatments we have studied, including cyclosporine [26], and narrow band ultraviolet B radiation [29]. Our group showed that TNFα blockade with etanercept reduced T cells, T17 cell products, and IL-17 induced genes, and that efficacy was associated with reduction in the IL-17 gene expression signature [67, 72]. Hence, for disease resolution, IL-17 must be switched off, implying that IL-17 signaling is required for disease presence. Three of the six known IL-17 ligands are overexpressed in psoriasis: IL-17A, IL-17F, and IL-17C. IL-17A and IL-17F bind to a heteromeric IL-17 receptor (IL-17R) composed of IL-17RA and IL-17RC, whereas IL-17C binds to an IL17RA/IL17RE complex [16]. There have been three anti-IL-17 agents tested in psoriasis, with slightly different targets. Secukinumab and ixekizumab selectively bind IL-17A [18, 28, 30, 33], whereas brodalumab inhibits the IL-17RA subunit [52, 53]. Hueber et al. first presented clinical improvement in psoriasis with secukinumab [28]. Studies with brodalumab and ixekizumab, in subjects with moderate-to-severe psoriasis, have shown rapid cellular and molecular responses to IL-17 blockade [30, 33, 52, 53]. Both brodalumab and ixekizumab appear to work quickly because there were significant improvements by 1–2 weeks. Furthermore, a large proportion of patients achieved PASI90 and PASI100 responses (75 % and 62 %, respectively, with brodalumab), indicating IL-17 pathway blockade eliminates an essential component of psoriasis. The extent to which IL-17 inhibitors reverse cellular, molecular, and clinical phenotypes of psoriasis was surprising, because multiple T-cell subtypes are coactivated in psoriasis. There are approximately 160 genes that are synergistically regulated by IL-17 and TNFα [8]. However, anti-IL-17A treatment of psoriasis reduced sixfold greater number of genes than anti-TNFα treatment at 2 weeks [30]. Also, genes synergistically regulated by IL-17 and TNFα were blocked to a higher magnitude by anti-IL-17A than anti-TNFα treatment. Synergism between IL-17 and IL-22 has also been shown [66], but may also exist with other IL-20 family cytokines (IL-19, IL-20, and IL-24) that bind a common receptor. Overall, this supports the crucial role of IL-17 in driving psoriasis pathogenesis and the complex synergistic effects of these pro-inflammatory cytokines.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


