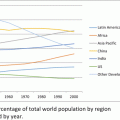
Fig. 5.1
The global investigative site landscape. Source: CenterWatch 2012 analysis of FDA-regulated investigators
In recent years, with changes in the healthcare arena, many physicians have considered getting involved in clinical research. The reasons for this have included alternative streams of revenue, fulfilling academic requirements, recognition among peers, specializing in a niche, getting published, and giving back to patients, the community, and the medical profession. Clinical trials are challenging, and require a great deal of training, organization, staff, and navigating a complex regulatory bureaucracy. Many investigators are first-time and one-time investigators. Annual surveys from the Tuft’s Center for the Study of Drug Development indicate a consistently high turnover rate for Physician Investigators.
According to Tuft’s data, in 2007; 26,000 Investigators registered with the FDA to conduct clinical trials; 85 % of the above registered physicians participated in only one trial (Fig. 5.2).
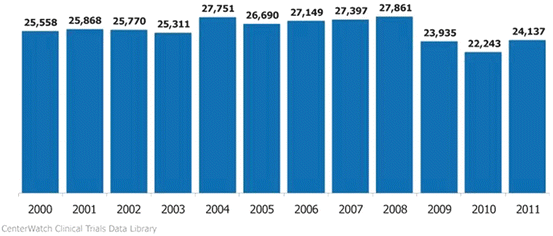
Fig. 5.2
Active unique investigators filing form 1572s worldwide. Source: FDA’s Bioresearch Monitoring Information System File (BMIS)
A lot of these physicians decided that research was not for them, or they did not have appropriate systems in place to support their research activities. While considering involvement in clinical research, several key factors should be taken into consideration:
Support staff: An essential staff member to have is a Clinical Research Coordinator (CRC). In my experience; nurses make the best coordinators. The CRC must handle a mix of clinical, administrative, business and marketing chores. Hiring a person with good personality is very important, because this person will become your liaison with Sponsors, Pharmaceutical and Biotech companies and CROs; and will be a leader for your patient recruitment and patient retention efforts. It would be a good idea to have your CRC be certified. Organizations like the ACRP and SOCRA grant certifications based on successful exams.
ACRP Certification, administered by the Academy of Clinical Research Professionals (the Academy), is the formal recognition of clinical research professionals who have met eligibility requirements and demonstrated proficiency of specific knowledge and job‐related skills by passing a standardized exam www.acrpnet.org.
Space: The space issue can be overlooked by a novice Investigator; some clinical trials will require that patients to stay at the Investigator’s site for more than a few hours. In addition to having regular practice exam rooms, you will need to provide an additional lounge or area so that the patient’s who are staying for a longer length of time will be comfortable and be able to have access to television, internet access, snacks, etc.
The FDA regulation requires storing investigational drugs in a secure space. In addition, the drug storage area should be temperature and preferably, humidity controlled. Investigational “scheduled” drugs must be stored in a double-locked cabinet or a locked cabinet that is in a secured locked room.
Make sure to plan for study coordinator space, sponsor monitor work space, and study records archive room. Please note that FDA regulation requires you to store study records for at least two (2) years after study closure and in some cases for a much longer time. As you grow your operation and take on more studies, you may consider an off-site medical archiving facility.
Equipment: Some clinical trials might require equipment that is not customary for your practice. You will need to have a centrifuge, locked refrigerator, and either a −20 °F or −80 °F freezer (or both) which all must be monitored daily. For your convenience I am including a sample of a temperature monitor log for your review. If you do not already have it in your practice, a defibrillator and medical emergency kit is a must.
Patient database: Historically the majority of clinical trials conducted in the United States do not enroll study subjects within the sponsor-anticipated enrollment period. I feel that one of the most important tasks before starting a trial is a careful evaluation of protocol-required patient population and making sure that you have a sufficient amount of these subjects within your database. At our site, we call this a protocol feasibility process. We not only assess the protocol-required subject population under diagnosis, but also review our electronic medical records against the study inclusion and exclusion criteria. In addition, we take into consideration the study duration, number of visits required, and duration of each visit. Consider your patient’s lifestyle, work situation, and age; they simply might not be willing to do the study because of time constrains.
Ask yourself a question: Will my patients benefit from participating in this trial? If the answer is yes, and you feel that you might be somehow limited with your patient database, consider reaching out to your colleagues in the medical community or advertising for the study subject. As we already mentioned above, all advertisement recruitment materials must be IRB approved. When considering approaching your colleagues for study subjects, please remember that incentive payments to healthcare professionals by Investigators for referral of study participants are known as referral fees. However, bear in mind that referral fees are not acceptable and may compromise the integrity of the research. In the (AMA) American Medical Association Code of Medical Ethics; Section 6.03 it is stated as follows: “Offering or accepting payment for referring patients to research studies (finder’s fees) are also unethical.”
Unless you have documented clinical research experience, securing the first clinical trial could be difficult. There are a couple of ways to go about this process.
Sponsors and CROs using databases to identify Investigators and sites for their upcoming clinical trials. I would recommend registering with each CRO and with DrugDev.org.
Another route, which is much easier, is to work with Site Management Organization (SMO), TMO or affiliate with a large dedicated research site in your region.
SMO and TMO will not only provide help with identifying clinical trials; but will help with managing some other very important aspects such as regulatory and IRB submissions, contract negotiation, marketing and advertising for your trials, and other important tasks.
Some SMOs might even help you with subject recruitment or place an experienced coordinator in your office.
There is a list of SMOs and dedicated research sites on the Center Watch website, www.centerwatch.com (Fig. 5.3).
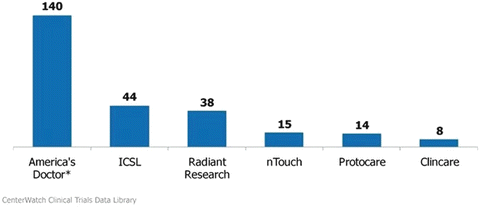
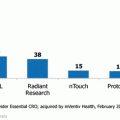
Fig. 5.3
Largest site management organizations in 2000 (number of sites in network). *Evolved into CRO service provider essential CRO; acquired by inVentiv Health, February 2010. Source: CenterWatch
5.2 Investigator-Initiated Trials
Some sponsors will reserve funds for small investigator-initiated studies, especially if they want to create good will at the site. Usually, these are conducted at large academic medical centers, with funding through grants, which are written by the investigators.
5.3 Training and Certification
SoCRA developed the Certified Clinical Research Professional Certification program to evaluate a CRPs knowledge, understanding, and application of the conduct of clinical investigations involving humans in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice (E6) (ICH/GCP), the United States Code of Federal Regulations (CFR), and the ethical principles that guide clinical research consistent with the principles of the Nuremberg Code, the Belmont Report, and the Declaration of Helsinki www.socra.org (Fig. 5.4).
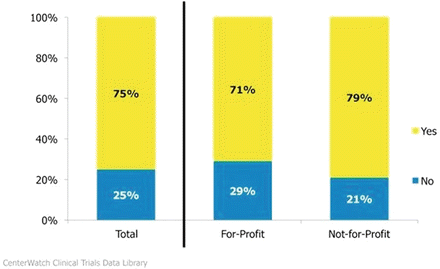
Fig. 5.4
Certification. Statistically significant at 95 %: none. Source: CenterWatch Clinical Research Coordinator Survey, 2012
5.4 SOPs
In clinical research, SOPs help define the Practice’s Research Department or Research Site standard practices and internal processes conducted to assure execution of research studies in accordance with Federal and State Regulations.
SOPs should guide research staff through a particular procedure and provide uniformity in the daily functions of the research department. The SOP should be written in a generic format to allow for easy implementation across a broad set of various aspects of multispecialty trials, but yet should specifically define procedures that can be followed without deviation.
The SOP should include the objective of the SOP, definition of key terms and acronyms. SOP must reference applicable guidances and regulations within the SOP, such as ICH E6 Good Clinical Practice and 21 CFR 50. The SOP should include the signature of the Research Administrator or Director with the date of approval.
SOPs should be reviewed at regular intervals (preferably annually) to reassess applicability of the policy.
Research staff should be educated and trained on SOPs. I suggest documenting the date when research staff have been appropriately trained and agree to comply with SOPs. Your staff should be monitored to ensure compliance and receive refresher training at regular intervals.
The following topics should be covered in your SOPs:
1.
SOP: Preparation, Issue, and Revisions
2.
Organizational Chart
3.
Master Study File and Record Retention
4.
Site QA and QC
5.
Subject Screening Procedures
6.
Informed Consent
7.
Study Initiation Visit
8.
Subject Study Visits
9.
Study Data Collection and Review
10.
Drug Storage, Inventory and Accountability
11.
Lab specimens Collection, Preparation and Shipment
12.
AE/SAE Documentation and Reporting
13.
Communication with and Reporting to IRB ( protocol departures, AE/SAE reporting)
14.
FDA Audit Preparation
15.
Study Close-out Visits
16.
Protection of Subject PHI and Site HIPAA Compliance
17.
Medical Emergency Procedures
18.
Staff Training
5.4.1 Source Document
Source document for a clinical trial might include, but is not limited to, the following: subject’s medical record, any protocol worksheets created for the study, and properly executed ICF. In addition, it may include other documents such as:
Subject Study diaries and questionnaires
EKG records
Laboratory reports
Letters and Memos
Original radiological films or Reports
Pathology slides
Tissue blocks
Drug dispensing log
Drug administration data (Medicine Administration Record—MAR)
Adverse events Serious Adverse Event log
5.4.2 Emergency Action Plan
During Katrina in 2005, there were 750 trials taking place at the Ochsner Clinic in New Orleans. Subjects had diseases from cancer to diabetes, from AIDS to COPD. All communication, including cell phones, and internet were disrupted. Only text messaging still worked. There was a huge loss to studies. Costs included patient contact information, data, and specimens. Furthermore, investigators left, 17 NIH funded labs at $5.7 M/year left LSU for institutions in other states. In 2012, Hurricane Sandy flooded New York City. Redundant generators providing power for NYU were unfortunately kept in the basement, where they flooded and malfunctioned. Patients had to be evacuated through stairwells because the elevators were not working. Some research records were destroyed.
Disasters happen, and can cripple research in any location. Backups can help protect your data. It’s a good idea to get as much contact information on subjects as you can (cell phone, email, and next of kin outside the area, if possible), store it in a secure place, and back it up off-site. Use radio communication with subjects after a disaster. You can call a news station and have them broadcast on the radio that your subjects are safe until authorities can arrive. Have contact information for local news and radio stations handy. Have remote storage and backup of all your data.
5.5 Feasibility
Feasibility is assessed at several levels. It is defined as the process of determining whether a trial can be conducted at your location given the parameters provided to you by your sponsor. Feasibility is very important to your sponsor, because nearly a 1/3 of the delay of clinical trials is due to subject enrollment. About 1/5 of investigative sites don’t enroll any subjects and about 1/3 of sites enroll only 5 % of total subjects. For sponsors, only 1/3 of the sites reliably enroll volunteers. You want to make sure you are in that 1/3 category. If you are, you will be in good company, and you will be invited back for future studies.
Feasibility at the program level is defined as an entire program, for example inflammatory skin disease, or cutaneous oncology. By definition, the scope of program level feasibility is broad. You have to decide if such a program has a reasonable chance of success in your area. For example, you may have a clinic in a small college town. The majority of your patients may be students, and the other faculty and residents of the town may be small in number but relatively young. A cutaneous oncology program focusing on skin cancers of the elderly may not be suitable for your research enterprise.
Feasibility at the study level has to do with your assessment of whether a study is doable. You want to analyze it from a clinical standpoint to see whether it is reasonable, but you also want to see if the regulatory, technical, and operational hurdles of the study are easily surmounted. Ask yourself if the protocol has any requirements which depart significantly from what you consider to be an acceptable study and clinical practices. Ask yourself if the regulatory burdens of the study allow it to be approved in a reasonable time frame, or if you will require time and resources to evaluate the study and get the approval of others in your research group, or other regulatory or administrative body. The study should require readily accessible technical tools, or supply them. Make sure there are no operational challenges to your efforts. For example, if your research center is in a community with several academic institutions or research centers, make sure that nearby sites are not being considered or haven’t been selected. This could hurt the sponsor and would hurt you, because nearby sites would be competing for volunteers from the same recruitment pool.
5.6 Protocol Feasibility
You will likely have to sign a confidentiality letter before you can look at a protocol. When you are reviewing a protocol, it is important for you to determine if it is feasible for your site. It is preferable to opt out of a study which is not suitable for you than to accept one and be unable to complete it because of foreseeable obstacles. Your ability to conduct and complete studies will build your reputation and attract more studies and sponsors. If you are regularly unable to fulfill study obligations, you may dim your prospects as an investigator.
First make sure you don’t have any competing trials. You will duplicate your efforts and dilute your enrollment. Ask yourself if you’ve done similar studies, and whether they were successful. If so, this is a good sign that the current trial is suitable for your site. The enrollment criteria should be clear and should be suitable for your patient population. Sometimes inclusion criteria are unclear. For example, if a protocol for psoriasis requires patients to enroll who are not taking steroid medication, without specifying how much steroid medication, for how long, and by what route (oral, topical, inhaled), you may inadvertently exclude or enroll the wrong subjects. Think about other factors which may hinder subject enrollment. For example, are subjects too young or too old? Does the study last too long (years?)? Are study visits too frequent, and would they interfere with the subjects’ daily lives? Is the dosing of the medication too frequent, or impractical (for example, after applying topical study drug, subjects must wait an hour before washing or wearing makeup)? The washout period allows subjects who are taking prohibited medications to discontinue the medications for a set period before being eligible for a trial. For example, subjects in a trial for atopic dermatitis may be allowed to start the trial after avoiding all topical steroids or topical immunomodulators for 4 weeks. Are there too many prohibitions on concomitant medications? Is the washout period too long, or too strict? Are there any procedures which cause undue discomfort to patients?
When reading the protocol, it’s important to ask yourself if criteria are vague, or unclear, or too strict. You can jot notes in the margin and bring up your questions during a site qualification visit or during the investigators meeting. Sponsors welcome Site/Investigator feedback. They want you to succeed and they want the study to succeed. It suggests an attentive and interested investigator. Often, the protocol will be amended based on Investigators feedback. The appointment times for visits should be suitable for your schedule and subjects’ schedules. A study with rigid visit windows over the holiday season may have limited enrollment or a high drop-out rate. A year-long study which requires that the same investigator evaluate a subject at each visit without provisions for substitute sub-investigators or co-investigators may not be feasible, or require a special subject retention plan. A study medication which requires occlusion with adhesive tape may not be suitable for subjects in a hot humid locale in the summer. If you find an obstacle in the study, try to think of suggestions to overcome it. A study drug for actinic keratosis which excludes patients with a history of skin cancer of any type could potentially be modified to include patients with a nonmelanoma skin cancer which has been successfully treated. You also need to determine if the sponsor has adequate resources and personnel to support you in the trial. If the trial requires specimens be shipped to a central laboratory, clearly marked shipping containers should be made available. If a protocol requires biopsy specimens to be snap frozen in liquid nitrogen and shipped on dry ice to a central laboratory future genetic analysis, be sure you get details. Look in the protocol for step-by-step instructions on obtaining and preparing the specimen. Check that you are supplied with all the necessary materials and reagents, including biopsy kits, sutures if necessary, specimen vials which are stable to snap freezing, liquid nitrogen and personal protective equipment for its handling, dry ice, insulated mail containers, added cost for transporting specimens, and clearances for shipping human tissue. It’s not good to find out midway in the study that a particular supply item is an out-of-pocket cost to you, or that your shipping container is not labeled or suitable for human specimens (Fig. 5.5).
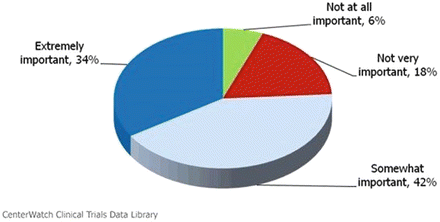
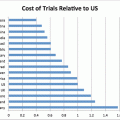
Fig. 5.5
How important is protocol design to your decision to participate in a clinical trial? Source: CenterWatch Survey of 1,329 investigative sites, 2010
Determine if you need specialized equipment. If this is something that needs to be purchased, is it in the budget? If this is something that you can lease or borrow from colleagues or a third party, will you be reimbursed for your costs? Ask where the study is conducted. Determine if the care is provided exclusively in your office, or your study site, or the hospital. Find out if you can see subjects in all locations, or if only certain sites can be used for part or all of the study. This is particularly important if you are working with co-investigators at multiple locations. Your sponsor, or the trial may insist that only one physical site be permitted for subject assessments. You may still be interested in such a trial, but you may have to be sure that travel times to the site and patient evaluation windows do not conflict. Ask if any special personnel are required to conduct the study. For example, you may be doing a trial on a biologic agent such as a tumor necrosis factor inhibitor, which is known to have potential cardiac toxicity, and may be required to do an electrocardiogram which is read by a board-certified cardiologist. Or, you may be required to administer a study drug intravenously. This will require trained and qualified staff. Ask if you will need to see subjects outside of standard clinic hours. For example, in phase I trials, some pharmacokinetic measurements need to be done over a 24-h time frame. Be sure you have the capability to deal with this exigency. Determine if, in your clinical opinion, adverse events, particularly severe adverse events are to be expected, or likely. This will add to your cost and time. You will need to budget accordingly. Alternatively, you may not be able to participate in a study of a medication fraught with burdensome side effects.
You also want to vet your sponsor or SMO/CRO. You want to research your sponsor’s track record, particularly in dermatology studies. You want to make sure they are adequately funded and have qualified and responsive personnel. You want to see that their protocol is well-thought out and well-written. If you find too many gaps or have to make too many suggestions and amendments just to get the protocol off the ground, of if your comments are brushed aside or disregarded, you may need to reconsider your collaboration with this Sponsor. You also want to gauge the sponsor’s long-term health. If you are audited by the FDA 2 years or 10 years after the study is completed, will the sponsor still be around?
5.7 Study Feasibility Checklist
Administrative support: from your practice manager, or your hospital or institution. If your institution doesn’t support you, you’ll have a difficult task succeeding in the study.
Subject recruitment/retention: How will you recruit subjects? Will you be able to get enough? Are the enrollment criteria reasonable or too restrictive? Does the sponsor help with recruitment? How sick is the subject population (greater risk of AE, SAE). Are there known risks for a similar class of drugs? How interesting is the study to you, or to your subjects (Botox)? Will you be able to retain your subjects? What compensation do they get? Is it fair or coercive? What about medication at the end of the trial?
You’ll need to screen many more subjects than you think. Typically 1 out of 16 potential subjects enrolls in a study, a rate of about 1 patient per month. The math is as follows: halve potential patients (lack of interest), halve again (childbearing), halve again (won’t or can’t consent), halve again (meet inclusion/exclusion criteria).
Keep track of your recruiting data and success rate. It will help you in future studies.
Knowing your screening-to-enrollment ratio helps you budget better. You need to know how many subjects you will have to screen in order to enroll one volunteer. You need to be aware how much time this will take. When you look at the inclusion and exclusion criteria, you have to determine if they are straightforward or time consuming, and budget accordingly.
If you share your misgivings about a study, the sponsor may relax the inclusion/exclusion criteria to help you meet your goal.
Attend investigators meeting (usually held before study initiation) where protocol is reviewed, and investigators have a chance to express their concerns and offer suggestions, and learn about the disease process being studied.
Designate a staff member with good interpersonal skill and good telephone manners to be your Subject Recruiter.
Compliance: is it too difficult, too uncomfortable, too tightly scheduled leading to high dropout rates? Will subjects have to miss school, work, vacation? Is dosing inconvenient, difficult, painful? Is there flexibility in the time schedule, or is it very rigid?
Personnel: do you have enough and do they have the right skills? If not, who will provide the training? How much time will training require, and will it take them away from other commitments? Do they understand their roles and responsibilities? Do they understand the ethics of clinical research? Do they understand confidentiality, HIPAA, FAA (Federal Aviation Administration) shipping regulations, OSHA regulations? Do they have the appropriate vaccinations and personal protective equipment?
PI: overall responsibility for the protocol, follows GCPs, federal and local regulations, ensuring proper subject consenting for the trial, properly delegate study-related task to well-trained and qualified site personnel, promptly report AEs and SAEs, serving as the patient care liaison between the sponsor and the IRB, makes medical assessments, provides guidance and oversees all members of the research team.
Study coordinator:
Helps determine study feasibility for the site
Assist the Investigator with performing all study-related tasks
Unless your site has a designated regulatory associate, your coordinator might be involved with handling and tracking study documents:
FDA 1572
CVs for all those listed in FDA 1572 and Financial Disclosures
IRB submission packet (protocol, consent, Investigator brochure)
Licenses (must be annually updated for each member and lab)
Maintaining the Regulatory binder
IRB and sponsor correspondence
Study guides and worksheets
Manages logistics
Schedules monitor visits, patient visits
Tracks patients study activities
Maintains study supplies
Assisting PI in subject selection and screening activities according to protocol inclusion/exclusion criteria
Takes part in subject consenting process
Performs non-MD study-related tasks
Coordinates monitor activities
Prepares case report forms (CRFs)
Makes sure all source docs are available
Assisting the sponsor monitor during their Site Visits
Works as a liaison between the lab, pharmacy, radiology, administration, IRB, dietary, housekeeping, etc.
Regulatory: can you meet IRB and consent requirements?
IRB packet contains the protocol, amendments, informed consent, and the Investigator Brochure.
Approval letter from IRB should include:
Name, address, and chair of IRB.
Name of contact person at IRB.
Name of the protocol and protocol identification number.
PI name and contact number.
Date of IRB approval.
Documents reviewed (protocol version number, amendments, etc.).
IRB decision (approval, not, any modifications).
IRB certification and list of members.
Signature of IRB chair.
Expiration date of approval.
Where the study may be conducted.
5.8 Regulations
5.8.1 Understanding the Regulations
In October of 2009 US Department of health and Human Services Food and Drug Administration issued a Guidance for Industry on Investigator Responsibilities—Protecting The Rights, Safety, and Welfare of Study Subjects.
The following excerpts from the guidance provide an overview of the responsibilities of a person (an Investigator as defined in 21 CFR 312(b)) who conduct a clinical investigation of a drug, biological product, or a medical device.
5.8.1.1 Overview of Investigator Responsibilities
In conducting clinical investigations of drugs, including biological products, under 21 CFR part 312 and of medical devices under 21 CFR part 812, the investigator is responsible for:
Ensuring that a clinical investigation is conducted according to the signed investigator statement for clinical investigations of drugs, including biological products, or agreement for clinical investigations of medical devices, the investigational plan, and applicable regulations
Protecting the rights, safety, and welfare of subjects under the investigator’s care
Controlling drugs, biological products, and devices under investigation (21 CFR 312.60, 21 CFR 812.100)
Investigators who conduct clinical investigations of drugs, including biological products, under 21 CFR Part 312, commit themselves to personally conduct or supervise the investigation. Investigators who conduct clinical investigations of medical devices, under 21 CFR Part 812, commit themselves to supervise all testing of the device involving human subjects. It is common practice for investigators to delegate certain study-related tasks to employees, colleagues, or other third parties (individuals or entities not under the direct supervision of the investigator). When tasks are delegated by an investigator, the investigator is responsible for providing adequate supervision of those to whom tasks are delegated. The investigator is accountable for regulatory violations resulting from failure to adequately supervise the conduct of the clinical study.
What Is Appropriate Delegation of Study-Related Tasks?
The investigator should ensure that any individual to whom a task is delegated is qualified by education, training, and experience (and state licensure where relevant) to perform the delegated task. Appropriate delegation is primarily an issue for tasks considered to be clinical or medical in nature, such as evaluating study subjects to assess clinical response to an investigational therapy (e.g., global assessment scales, vital signs) or providing medical care to subjects during the course of the study. Most clinical/medical tasks require formal medical training and may also have licensing or certification requirements. Licensing requirements may vary by jurisdiction (e.g., states, countries). Investigators should take such qualifications/licensing requirements into account when considering delegation of specific tasks. In all cases, a qualified physician (or dentist) should be responsible for all trial-related medical (or dental) decisions and care.
During inspections of investigation sites, FDA has identified instances in which study tasks have been delegated to individuals lacking appropriate qualifications. Examples of tasks that have been inappropriately delegated include:
Screening evaluations, including obtaining medical histories and assessment of inclusion/exclusion criteria
Physical examinations
Evaluation of adverse events
Assessments of primary study endpoints
Obtaining informed consent
The investigator is responsible for conducting studies in accordance with the protocol (see 21 CFR 312.60, Form FDA-1572, 21 CFR 812.43 and 812.100). In some cases a protocol may specify the qualifications of the individuals who are to perform certain protocol-required tasks (e.g., physician, registered nurse), in which case the protocol must be followed even if state law permits individuals with different qualifications to perform the task (see 21 CFR 312.23(a)(6) and 312.40(a)(1)). For example, if the state in which the study site is located permits a nurse practitioner or physician’s assistant to perform physical examinations under the supervision of a physician, but the protocol specifies that physical examinations must be done by a physician; a physician must perform such exams.
The investigator should maintain a list of the appropriately qualified persons to whom significant trial-related duties have been delegated. This list should also describe the delegated tasks and identify the training that individuals have received that qualifies them to perform delegated tasks.
What Is Adequate Supervision of the Conduct of an Ongoing Clinical Trial?
For each study site, there should be a distinct individual identified as an investigator who has supervisory responsibility for the site. Where there is a subinvestigator at a site, that individual should report directly to the investigator for the site (i.e., the investigator should have clear responsibility for evaluating the subinvestigator’s performance and the authority to terminate the subinvestigator’s involvement with the study) and the subinvestigator should not be delegated the primary supervisory responsibility for the site.
The investigator should have sufficient time to properly conduct and supervise the clinical trial. The level of supervision should be appropriate to the staff, the nature of the trial, and the subject population. In FDA’s experience, the following factors may affect the ability of an investigator to provide adequate supervision of the conduct of an ongoing clinical trial at the investigator’s site:
Inexperienced study staff
Demanding workload for study staff
Complex clinical trials (e.g., many observations, large amounts of data collected)
Large number of subjects enrolled at a site
A subject population that is seriously ill
Conducting multiple studies concurrently
Conducting a study from a remote (e.g., off-site) location
Conducting a study at multiple sites under the oversight of a single investigator, particularly where those sites are not in close proximity
The investigator should develop a plan for the supervision and oversight of the clinical trial at the site. Supervision and oversight should be provided even for individuals who are highly qualified and experienced.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


